
КАЙЕНДРА
-
Швейцарія Новартіс Оверсіз Інвестментс АГDosage form: таблетки, вкриті плівковою оболонкою, по 0,25 мг по 12 таблеток у блістері; по 1 блістеру в пеналі; по 1 пеналу у картонінй коробціComposition: 1 таблетка, вкрита плівковою оболонкою, містить 0,25 мг сипонімоду (у вигляді сипонімоду фумарової кислоти);Medicinal dispensing terms: за рецептомСertificate number: UA/20733/01/01ATS code: L04AA42Expiry: 18 місяців.
-
Швейцарія Новартіс Оверсіз Інвестментс АГDosage form: таблетки, вкриті плівковою оболонкою, по 2 мг; по 14 таблеток у блістері; по 2 блістери у картонній коробціComposition: 1 таблетка, вкрита плівковою оболонкою, містить 2 мг сипонімоду (у вигляді сипонімоду фумарової кислоти);Medicinal dispensing terms: за рецептомСertificate number: UA/20733/01/02ATS code: L04AA42Expiry: 18 місяців.
ІНСТРУКЦІЯ
для медичного застосування лікарського засобу
КАЙЕНДРА
(KIENDRA)
Склад:
діюча речовина: сипонімод;
1 таблетка, вкрита плівковою оболонкою, містить 0,25 мг сипонімоду (у вигляді сипонімоду фумарової кислоти);
1 таблетка, вкрита плівковою оболонкою, містить 2 мг сипонімоду (у вигляді сипонімоду фумарової кислоти);
допоміжні речовини: лактоза, моногідрат; целюлоза мікрокристалічна, кросповідон, гліцерол дибегенат, кремнію діоксид колоїдний безводний, спирт полівініловий, титану діоксид (Е 171), заліза оксид жовтий (Е 172) – лише для дозування 2 мг, заліза оксид червоний (Е 172), заліза оксид чорний (Е 172) – лише для дозування 0,25 мг, тальк, лецитин (Е 322), ксантанова камедь.
Лікарська форма. Таблетки, вкриті плівковою оболонкою.
Основні фізико-хімічні властивості:
таблетки, вкриті плівковою оболонкою, по 0,25 мг: блідо-червоні, без риски, круглі, двоопуклі таблетки, вкриті плівковою оболонкою, зі скошеними краями, з тисненням логотипу «
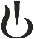 » з
одного боку та «T» з іншого;
» з
одного боку та «T» з іншого;
таблетки, вкриті плівковою оболонкою, по 2 мг: блідо-жовті, без риски, круглі, двоопуклі таблетки, вкриті плівковою оболонкою, зі скошеними краями, з тисненням логотипу «
» з
одного боку та «ІІ» з іншого.
Фармакотерапевтична група. Антинеопластичні та імуномодулюючі засоби. Імуносупресанти. Селективні імуносупресанти. Код АТХ L04A A42.
Фармакологічні властивості.
Фармакодинаміка
Сипонімод являє собою модулятор сфінгозин-1-фосфатних рецепторів (S1P). Сипонімод селективно зв’язується з двома з п’яти рецепторів S1P, пов’язаних з G-білками (GPCR), а саме з S1P1 і S1P5. Як функціональний антагоніст рецепторів S1P1 на поверхні лімфоцитів сипонімод перешкоджає їх виходу з лімфатичних вузлів. Це призводить до зниження рециркуляції Т-лімфоцитів в центральну нервову систему (ЦНС) і, таким чином, обмежує розвиток запалення ЦНС. Сипонімод проникає крізь гематоенцефалічний бар’єр. Сипонімод не чинить тривалої дії на ефекторні Т-клітини пам’яті в периферичній тканині та крові та не погіршує активацію лімфоцитів.
У дослідженнях на тваринах був продемонстрований прямий вплив сипонімоду на нервові клітини: через S1P1 – на астроцити та через S1P5 – на олігодендроцити. На моделі мишей експериментального аутоімунного енцефаломієліту також був продемонстрований незалежний від впливу на лімфоцити прямий нейропротекторний ефект сипонімоду, що застосовується централізовано (шляхом інтрацеребровентрикулярних інфузій).
Імунна система
Препарат Кайендра спричиняє дозозалежне зниження кількості лімфоцитів у периферичній крові протягом 6 годин після прийому першої дози внаслідок зворотної секвестрації лімфоцитів у лімфоїдній тканині.
На тлі тривалого щоденного прийому кількість лімфоцитів продовжує знижуватися, досягаючи найменшого серединного значення (90 % ДІ), яке приблизно дорівнює 0,560 (0,271–1,08) клітин/нл, що становить 20–30 % від вихідного рівня у типових пацієнтів неяпонського походження з вторинно-прогресуючим розсіяним склерозом (ВПРС) з генотипом*1*1 або *1*2 гена CYP2C9. Низька кількість лімфоцитів зберігається при тривалому щоденному прийомі.
У переважної більшості (90 %) пацієнтів з ВПРС число лімфоцитів повертається до нормального рівня протягом 10 днів після припинення терапії. Остаточний ефект зниження кількості лімфоцитів в периферичній крові може зберігатися протягом 3–4 тижнів після прийому останньої дози препарату Кайендра.
Кардіоелектрофізіологія
Частота і ритм серцевих скорочень
Прийом препарату Кайендра викликає тимчасове зменшення ЧСС і атріовентрикулярної провідності на початку лікування (див. розділ «Побічні реакції»), що за механізмом дії пов’язано з активацією пов’язаних з G-білком калієвих каналів внутрішнього випрямлення за допомогою стимуляції рецепторів S1P1, наслідком чого є гіперполяризація клітин і зниження збудливості. Внаслідок функціонального антагонізму сипонімоду з рецепторами S1P1 при початковому підборі дози виникає послідовна десенсибілізація каналів GIRK, яка відмічається до досягнення підтримуючої дози.
Потенціал до пролонгації інтервалу QT
Вплив терапевтичних (2 мг) та супратерапевтичних (10 мг) доз сипонімоду на реполяризацію серця вивчався в спеціальному QT-дослідженні. Результати не підтвердили наявність аритмогенного потенціалу, пов’язаного з пролонгацією інтервалу QT при прийомі сипонімоду, оскільки через 3 години після прийому дози сипонімоду збільшувалося середнє плацебо-скориговане значення QTcF (ΔΔQTcF) з поправкою на початкове значення більш ніж на 5 мс, середній максимальний ефект становив 7,8 мс при прийомі дози 2 мг і 7,2 мс при дозі 10 мг. Верхня межа одностороннього 95 % довірчого інтервалу для ΔΔQTcF у всіх тимчасових точках залишалася нижче 10 мс. Категоріальний аналіз не виявив випадків подовження інтервалу QTc > 480 мс на тлі лікування, випадків подовження інтервалу QTc на > 60 мс від вихідного рівня і випадків збільшення скоригованого або нескоригованого значення QT/QTc > 500 мс.
Функція легень
Прийом однієї або декількох доз препарату Кайендра протягом 28 днів не супроводжувався клінічно значущим збільшенням опору дихальних шляхів при проведенні оцінки за величиною ОФВ1 і миттєвої об’ємної швидкості видиху 25–75 % (МОС 25–75) від форсованої життєвої ємності легень (ФЖЕЛ). При одноразовому прийомі нетерапевтичної дози (> 10 мг) зафіксована незначна тенденція до зниження ОФВ1. Прийом декількох доз призводив до легких або помірних змін ОФВ1 і МОС 25–75 % незалежно від величини дози і часу доби, які не супроводжувалися клінічними проявами збільшення опору дихальних шляхів.
Клінічна ефективність
Ефективність препарату Кайендра вивчалась у дослідженні ІІІ фази з оцінкою дози 2 мг препарату Кайендра один раз на добу у пацієнтів з ВПРС. Дослідження ІІ фази дозування у пацієнтів з РРРС продемонструвало дозозалежне зменшення запальних уражень на МРТ і виявило, що 2 мг препарату Кайендра забезпечує майже максимальний ефект.
Дослідження A2304 (EXPAND) за участю пацієнтів з ВПРС
Дослідження A2304 являло собою рандомізоване, подвійно сліпе, плацебо-контрольоване, кероване за тривалістю подій і періоду подальшого спостереження дослідження III фази за участю пацієнтів з ВПРС з документально підтвердженими ознаками прогресування в попередні 2 роки за відсутності або незалежно від рецидивів, відсутності доказів рецидиву протягом 3 місяців до включення в дослідження і з медіанним балом за Розширеною шкалою оцінки ступеня інвалідизації (EDSS) від 3,0 до 6,5 на момент включення в дослідження.
Вихідна медіана EDSS становила 6,0. Пацієнти віком понад 61 рік не були включені в дослідження. Що стосується активності захворювання, ознаки, характерні для запальної активності в ВПРС, можуть бути пов’язані з рецидивом або з виконанням візуалізаційних досліджень (тобто Gd-накопичувальні вогнища на T1-зважених зображеннях або активні [нові або накопичувальні] вогнища на T2-зважених зображеннях).
Пацієнти були рандомізовані у співвідношенні 2:1 в групи прийому препарату Кайендра у дозі 2 мг один раз на добу або плацебо. Клінічні оцінки проводили під час скринінгу, кожні 3 місяці і під час рецидиву. Оцінки МРТ проводили під час скринінгу і кожні 12 місяців.
Первинною кінцевою точкою дослідження був час до 3-місячного підтвердженого прогресування інвалідності (CDP), яке визначається як збільшення не менш ніж на 1 бал від початкового значення EDSS (збільшення на 0,5 бала для пацієнтів з вихідним значенням EDSS 5,5 або більше), що підтримувалось протягом 3 місяців. Основними вторинними кінцевими точками були час до 3-місячного підтвердженого погіршення не менш ніж на 20 % від початкового значення тесту 25-футової ходьби з урахуванням часу (T25W) і зміна об’єму ураження на T2-зваженому зображенні в порівнянні з вихідним значенням. Додаткові вторинні кінцеві точки включали час до 6-місячного CDP, відсоткову зміну об’єму мозку і показники активності запального захворювання (частота рецидиву за рік; ураження, оцінені на МРТ). Пошуковою кінцевою точкою дослідження була зміна швидкості когнітивної обробки, виявлена за результатами тесту на зіставлення символів і цифр.
Тривалість дослідження була різною для окремих пацієнтів (медіана тривалості дослідження становила 21 місяць, діапазон від 1 дня до 37 місяців).
В дослідженні було рандомізовано1651 пацієнта в групу прийому препарату Кайендра 2 мг (N = 1105) або плацебо (N = 546); 82 % пацієнтів, які отримували препарат Кайендра, і 78 % пацієнтів, які отримували плацебо, завершили дослідження. На початок дослідження медіана віку становила 49 років, середня тривалість захворювання – 16 років, а середній бал за шкалою EDSS – 6,0. У 64 % пацієнтів не було рецидивів протягом 2 років до початку дослідження, а у 76 % не відмічалось гадоліній (Gd)-накопичувальних вогнищ на початковому МРТ-скануванні. 78 % пацієнтів раніше отримували терапію з приводу РС.
Час початку 3-місячного і 6-місячного підтвердженого прогресування інвалідності було значно відстрочено для сипонімоду зі зниженням ризику 3-місячного CDP на 21 % в порівнянні з плацебо (відношення ризиків [HR] 0,79, p = 0,0134) і зниженням ризику 6-місячного CDP на 26 % в порівнянні з плацебо (HR 0,74, р = 0,0058).
Результати цього дослідження наведено у таблиці 1 та на рис. 1 та 2.
Таблиця 1
Клінічні та МРТ результати дослідження A2304
|
Кінцеві точки |
A2304 (EXPAND) |
|
|
2 мг сипонімоду (n = 1099) |
Плацебо (n = 546) |
|
|
Клінічні кінцеві точки |
||
|
Основна клінічна точка ефективності Відсоткова частка пацієнтів з підтвердженим прогресуванням інвалідизації за 3 місяці (основна кінцева точка ) |
26,3 % |
31,7 % |
|
Зниження ризику1 |
21 % (p = 0,0134) |
|
|
Відсоткова частка пацієнтів з підтвердженим 20 % збільшенням значення тесту 25-футової ходьби за 3 місяці |
39,7 % |
41,4 % |
|
Зниження ризику1 |
6 % (p = 0,4398 |
|
|
Відсоткова частка пацієнтів з підтвердженим прогресуванням інвалідизації за 6 місяців |
19,9 % |
25,5 % |
|
Зниження ризику1 |
26 % [(p = 0,0058)]6 |
|
|
Частота рецидиву за рік (ARR) |
0,071 |
0,152 |
|
Зниження частоти2 |
55 % [(p < 0,0001)]6 |
|
|
Кінцеві точки МРТ |
||
|
Зміна об’єму ураження від вихідного T2-зважених зображень (мм3)3 |
+184 мм3 |
+879 мм3 |
|
Різниця в зміні об’єму ураження T2-зважених зображень |
-695 мм3 (p < 0,0001)7 |
|
|
Відсоткова зміна об’єму головного мозку від початкового значення (95 % ДІ)3 |
-0,497 % |
-0,649 % |
|
Різниця у відсотковій зміні об’єму головного мозку |
0,152 % [(p = 0,0002)]6 |
|
|
Середня сумарна кількість Gd-накопичувальних вогнищ на T1-зважених зображеннях (95 % ДІ)4 |
0,081 |
0,596 |
|
Зниження частоти |
86 % [(p < 0,0001)]6 |
|
|
Відсоткова частка пацієнтів з погіршенням на 4 бали, виявленим за результатами тесту на зіставлення символів і ціфр5 |
16,0 % |
20,9 % |
|
Зниження ризику1 |
25 % [(p = 0,0163)]6 |
|
|
1 З моделювання Кокса для часу до прогресування. 2 З моделі для випадків рецидиву. 3 Середнє значення за 12 і 24 місяці. 4 До 24 місяців. 5 Підтверджено через 6 місяців. 6 [Номінальне p-значення для кінцевих точок, які не включені в багаторівневе тестування і не скориговані на множинність]. 7 Непідтверджене p-значення; процедура багаторівневого тестування припинена до досягнення кінцевої точки. |
||
|
Час до 3-місячного CDP в порівнянні з плацебо |
Час до 6-місячного CDP в порівнянні з плацебо |
|
Відсоткова частка пацієнтів з 6-місячним підтвердженим прогресуванням інвалідизації |
|
Сипонімод Плацебо |
|
Сипонімод Плацебо |
|
Сипонімод |
|
Плацебо |
|
Кількість пацієнтів з ризиком |
|
Кількість пацієнтів з ризиком |
|
Місяць дослідження |
|
Місяць дослідження |
|
Відношення ризиків: 0,74, р = 0,0058; 95 % ДІ: 0,60, 0,90); зниження ризику: 26 % |
|
Відношення ризиків: 0,79, р = 0,0134; 95 % ДІ: 0,65, 0,95); зниження ризику: 21 % |
|
Відсоткова частка пацієнтів з 3- місячним підтвердженим прогресуванням інвалідизації |
Рис. 1. Пацієнти з 3- та 6-місячним підтвердженим прогресуванням інвалідизації на підставі розширеної шкали оцінки ступеня інвалідизації – кривої Каплана – Мейєра (повна вибірка пацієнтів для аналізу, дослідження A2304)
Результати дослідження продемонстрували послідовне зниження ризику під час 3- і 6-місячного CDP при прийомі препарату Кайендра в порівнянні з плацебо в підгрупах, сформованих за статтю, віком, попередньою терапією розсіяного склерозу, активністю рецидиву до проведення дослідження, вихідною активністю захворювання за результатами МРТ, тривалістю захворювання і вихідними показниками інвалідності.
Препарат Кайендра продемонстрував позитивний ефект під час тесту на зіставлення символів і цифр (SDMT). Зміна порівняно з вихідними значеннями була стабільною або кращою при прийомі препарату Кайендра та гіршою при прийомі плацебо зі значною різницею між групами: 1,1 бала через 12 місяців (p = 0,0132) та 2,3 бала через 24 місяці (p = 0,0002). В експериментальному дослідженні препарат Кайендра знижував ризик підтвердженого 4-бального погіршення в тесті SDMT через 6 місяців на 25 % (p = 0,0163) порівняно з плацебо. 4-бальне погіршення виявилось клінічно значущим.
У підгрупі пацієнтів (47,1 %, n = 779) з активним захворюванням (що визначається як пацієнти з рецидивом за 2 роки до дослідження та/або з наявністю Gd-накопичувальних вогнищ на T1-зважених зображеннях на вихідному рівні) вихідні характеристики були аналогічні таким у загальній популяції. На початку дослідження медіана віку становила 47 років, середня тривалість захворювання – 15 років, а середній бал за шкалою EDSS – 6,0 (див. розділ «Фармакокінетика»). Час до початку 3-місячного і 6-місячного CDP було значно відстрочено у пацієнтів з активним захворюванням, які приймали сипонімод: на 31 % в порівнянні з пацієнтами, які приймали плацебо (відношення ризиків [HR] 0,69; 95 % ДІ: 0,53, 0, 91) і на 37 % в порівнянні з пацієнтами, які приймали плацебо (HR 0,63; 95 % ДІ: 0,47, 0,86) відповідно. Частота ARR (підтверджених рецидивів) знижувалася на 46 % (співвідношення ARR 0,54; 95 % ДІ: 0,39, 0,77) в порівнянні з плацебо. Відносне зниження частоти сумарного числа Gd-накопичувальних вогнищ на T1-зважених зображеннях за 24 місяці склало 85 % (співвідношення ризиків 0,155; 95 % ДІ: 0,104, 0,231) в порівнянні з плацебо. Відмінності в зміні обсягу вогнищ на Т2-зважених зображеннях і у відсотках зміни об’єму головного мозку (в середньому за 12 і 24 місяці) в порівнянні з плацебо становили -1163 мм3 (95 % ДІ: 1484, 843 мм3) і 0,141% ( 95% ДІ: 0,020, 0,261 %) відповідно.
У підгрупі пацієнтів (n = 827) без ознак або симптомів активності захворювання (що визначається як пацієнти без рецидиву за 2 роки до дослідження та без наявності помірних накопичувальних вогнищ на контрастних T1-зображеннях на вихідному рівні) вплив на 3-місячне та 6-місячне підтверджене прогресування інвалідизації був незначним (зниження ризиків становило 7 % та 13 % відповідно).
|
Час до 3-місячного CDP в порівнянні з плацебо (первинна кінцева точка) |
Час до 6-місячного CDP в порівнянні з плацебо |
|
Відсоткова частка пацієнтів з 6-місячним підтвердженим прогресуванням інвалідизації |
|
Відношення ризиків: 0,69, (95% ДІ: 0,453, 0,91); зниження ризику: 31% |
|
Сипонімод |
|
Плацебо |
|
Сипонімод Плацебо |
|
Сипонімод Плацебо |
|
Кількість пацієнтів з ризиком |
|
Кількість пацієнтів з ризиком |
|
Відношення ризиків: 0,63, (95% ДІ: 0,47, 0,86); зниження ризику: 37% |
|
Місяць дослідження |
|
Місяць дослідження |
|
Відсоткова частка пацієнтів з 3-місячним підтвердженим прогресуванням інвалідизації |
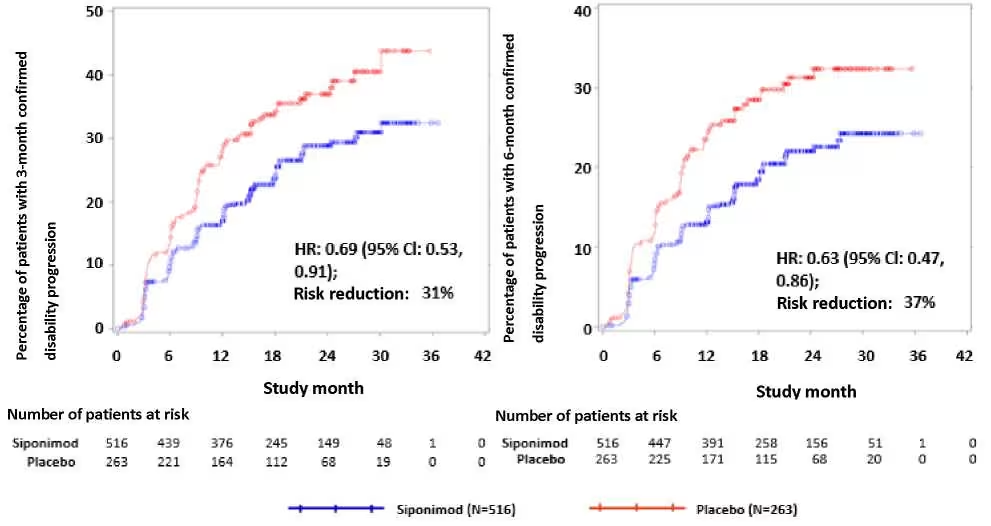
Рис. 2. Пацієнти з 3- та 6-місячним підтвердженим прогресуванням інвалідизації на підставі розширеної шкали оцінки ступеня інвалідизації – кривої Каплана – Мейєра – підгрупа з активним запальним захворюванням (повна вибірка пацієнтів для аналізу, дослідження A2304)
Фармакокінетика
Абсорбція
Час (Tmax) до досягнення максимальної концентрації сипонімоду в плазмі крові (Cmax) після багаторазового перорального застосування становить близько 4 годин (діапазон від 2 до 12 годин). Абсолютна біодоступність сипонімоду при прийомі всередину становить близько 84 %. При прийомі сипонімоду в дозі 2 мг один раз на добу протягом 10 днів середнє значення Cmax на десятий день становило 30,4 нг/мл, а середнє значення AUCtau 558 г*нг/мл спостерігалося на десятий день. Рівноважний стан був досягнутий приблизно через 6 днів після багаторазового прийому сипонімоду один раз на добу.
Прийом їжі не впливав на системну експозицію сипонімоду (Cmax та AUC). Отже, препарат Кайендра можна застосовувати незалежно від прийому їжі.
Розподіл
Сипонімод розподіляється в тканинах, середній об’єм розподілу становить 124 л. Частка сипонімоду в плазмі крові у людини становить 68 %. Дослідження на тваринах показують, що сипонімод легко проникає через гематоенцефалічний бар’єр. Ступінь зв’язування сипонімоду з білками плазми крові у здорових добровольців і у пацієнтів з порушенням функції печінки і нирок становить > 99,9 %.
Метаболізм
Сипонімод значною мірою метаболізується, переважно цитохромом CYP2C9 (79,3 %) і меншою мірою CYP3A4 (18,5 %).
Не очікується, що фармакологічна активність основних метаболітів М3 і М17 сприятиме клінічному ефекту і безпеці застосування сипонімоду у людини.
Виведення
У пацієнтів з РС оцінений системний кліренс (CL/F) становив 3,11 л/год. Фармакокінетично важливий період напіввиведення становить приблизно 30 годин.
Сипонімод виводиться з системного кровообігу в основному за рахунок обміну речовин і подальшої екскреції з жовчю/фекаліями. Близько 86,7 % дози сипонімоду виводиться з калом, 9,2 % – в незмінному вигляді. Лише незначна кількість дози виводиться із сечею (3,6 %). Незмінений сипонімод не був виявлений в сечі.
Середні періоди напіввиведення метаболітів M17 та M3 сипонімоду після перорального застосування становлять близько 155 год та 30 год відповідно.
Лінійність/нелінійність
Концентрація сипонімоду збільшується пропорційно дозі, що спостерігається після кількох прийомів сипонімоду у дозі від 0,3 до 20 мг один раз на добу.
Концентрація сипонімоду в рівноважному стані в 2–3 рази перевищує концентрацію після прийому першої дози і досягається приблизно через 6 днів при прийомі один раз на добу. Для досягнення клінічної терапевтичної дози 2 мг необхідний шестиденний період фази титрування, після чого потрібно додатково 4 дні прийому для досягнення рівноважної концентрації в плазмі крові.
Фармакокінетика в особливих групах пацієнтів
Генотип CYP2C9
Генотип CYP2C9 має суттєвий вплив на метаболізм сипонімоду.
Лікування препаратом Кайендра протипоказано пацієнтам, гомозиготним за алелем CYP2C9*3 (генотип CYP2C9*3*3) (див. розділи «Протипоказання», «Особливості застосування» та «Взаємодія з іншими лікарськими засобами та інші види взаємодій»). У таких пацієнтів застосування препарату Кайендра призводить до значного підвищення рівня сипонімоду в плазмі. Рекомендована підтримуюча доза препарату Кайендра становить 1 мг на добу для пацієнтів із генотипом CYP2C9*2*3 або *1*3, щоб уникнути підвищеного впливу сипонімоду (див. розділ «Спосіб застосування та дози»).
Існують інші, менш поширені поліморфізми CYP2C9. Фармакокінетика сипонімоду не була оцінена у носіїв цих генотипів. Деякі з цих поліморфізмів, зокрема алелі *5, *6, *8 і *11, також пов’язані зі зниженням або втратою функції ферменту (див. також розділи «Особливості застосування» і «Взаємодія з іншими лікарськими засобами та інші види взаємодій»).
Після прийому одноразової дози сипонімоду 0,25 мг AUCinf та AUClast були приблизно у 2–4 рази вищими у пацієнтів із генотипом *2*3 гена CYP2C9 та генотипом *3*3 гена CYP2C9 відповідно, хоча відмічалось тільки незначне збільшення – на 21 % та 16 % відповідно – порівняно зі швидкими метаболізаторами (CYP2C9 *1*1). Середній період напіввиведення був подовжений у носіїв генотипу*2*3 гена CYP2C9 та генотипу *3*3 гена CYP2C9 (51 год та 126 год відповідно).
Очевидний системний кліренс (CL/F) 3,11 л/год оцінювався у екстенсивному метаболізаторі CYP2C9 (генотип *1*1 гена CYP2C9 та генотип *1* 2 гена CYP2C9) у пацієнтів з ВПРС після багаторазового перорального введення сипонімоду. CL/F становить 2,5, 1,9, 1,6 та 0,9 л/год у пацієнтів із генотипом *2*2 гена CYP2C9, генотипом *1*3 гена CYP2C9, з генотипом *2*3 гена CYP2C9 та генотипом *3*3 гена CYP2C9 відповідно. Отримане збільшення AUC сипонімоду становило 25, 61, 91 та 285 % у пацієнтів із генотипом *2*2 гена CYP2C9, генотипом *1*3 гена CYP2C9, з генотипом *2*3 гена CYP2C9 та генотипом *3*3 гена CYP2C9 відповідно, порівняно з пацієнтами, які мають генотип *1*1 гена CYP2C9. Оскільки очевидний кліренс, оцінений для пацієнтів з генотипом *1*2 гена CYP2C9, був порівнянним з таким у пацієнтів з генотипом *1*1 гена CYP2C9, аналогічний вплив сипонімоду очікується для обох генотипів.
Пацієнти з порушенням функції нирок
Корекція дози сипонімоду для пацієнтів зі слабким, помірним та тяжким порушенням функції нирок не потрібна. Значення середнього періоду напіввиведення і Cmax сипонімоду (загального і незв’язаного) можна було порівняти у пацієнтів з тяжким порушенням функції нирок та у здорових осіб. Значення загальної та незв’язаної AUC були незначно збільшені (на 23–33 %) у порівнянні зі значеннями у здорових пацієнтів. Вплив термінальної стадії ниркової недостатності або гемодіалізу на фармакокінетику сипонімоду не вивчався. Через високе зв’язування сипонімоду з білками плазми (понад 99,9 %) малоймовірно, що гемодіаліз може змінювати концентрацію загального і незв’язаного сипонімоду, тому не очікується, що виникне потреба в корекції дози.
Пацієнти з порушенням функції печінки
Пацієнтам з порушенням функції печінки корекція дози сипонімоду не потрібна. Значення AUC незв’язаного сипонімоду на 15–50 % вищі у пацієнтів з помірним та тяжким порушенням функції печінки, ніж у пацієнтів з нормальною функцією печінки, при прийомі досліджуваної дози 0,25 мг. Середній період напіввиведення сипонімоду залишався незміненим у пацієнтів з порушенням функції печінки.
Пацієнти літнього віку
У клінічні дослідження були включені пацієнти віком до 61 років (див. розділ «Спосіб застосування та дози»).
Стать
Стать не впливає на фармакокінетику сипонімоду.
Расова/етнічна група
Фармакокінетичні параметри одноразової дози у пацієнтів-японців і пацієнтів європеоїдної раси не розрізнялися, що вказує на відсутність впливу етнічних особливостей на фармакокінетику сипонімоду.
Дані доклінічних досліджень
Доклінічний профіль безпеки сипонімоду оцінювали у дослідженнях токсичності одноразових та повторних доз на мишах (до 13 тижнів), щурах (до 26 тижнів) та мавпах (до 52 тижнів). Дозолімітуючими токсичними ефектами були нефротоксичність у мишей, підвищення маси тіла у щурів та несприятливий вплив на ЦНС та шлунково-кишковий тракт у мавп. Основними органами-мішенями токсичності, виявленими під час гістопатологічних досліджень у гризунів, були легені, печінка, щитовидна залоза, нирки та матка/піхва. Вплив на м’язи та шкіру спостерігався у окремих мавп.
NOAEL у щурів встановлювали на рівні 50 та 15 мг/кг/добу відповідно для самців та самок, а у мавп – 10 мг/кг/добу для обох статей. Межі безпеки на підставі AUC для системних ефектів (фактор 171) були розраховані на основі підтримуючої дози 2 мг/добу.
Генотоксичність та канцерогенність
Тести in vitro для оцінки генотоксичного потенціалу (тест на зворотні мутації, мікроядерний тест та аналіз хромосомних аберацій у культурі лімфоцитів людини) та мікроядерний тест in vivo на щурах не виявили жодного генотоксичного потенціалу сипонімоду.
У дослідженнях канцерогенності сипонімод індукував виникнення лімфоми, гемангіоми та гемангіосаркоми у мишей, тоді як у самців щурів були виявлені фолікулярна аденома і карцинома щитовидної залози. Такі результати щодо пухлин були розцінені або як специфічні для мишей, або як обумовлені метаболічними адаптаціями печінки у щурів. Клінічне значення для людини невідоме.
Концентрація сипонімоду в плазмі крові (AUC) при найнижчій досліджуваній дозі у мишей приблизно в 29 разів вища, ніж при рекомендованій дозі 2 мг у людини.
Фертильність та репродуктивна токсичність
Сипонімод не впливав на фертильність самців і самок щурів аж до найвищої досліджуваної дози, що призводить до встановлення межі безпеки, яка в 16 разів перевищує значення, визначене на підставі системної експозиції у людей (AUC) при добовій дозі 2 мг. Вплив на репродуктивні органи у щурів та мавп не спостерігався після тривалого введення.
Репродуктивні дослідження та дослідження розвитку на вагітних щурах та кролях продемонстрували індуковану сипонімодом ембріотоксичність і фетотоксичність у щурів та кролів, а також тератогенність у щурів. Підвищена частота постімплантаційних втрат та аномалій плода (зовнішніх, сечостатевих та скелетних) у щурів та ембріо-фетальних смертей, викиднів та змін у плода (скелетних та вісцеральних) у кролів спостерігалась після пренатального впливу сипонімоду в дозі, що відповідала рекомендованій добовій дозі 2 мг для людини.
Рівні експозиції у щурів та кролів у випадках, коли не повідомлялося про вади розвитку або ембріофетальну смерть, були субклінічними. Це свідчить про те, що при підтримуючій добовій дозі 2 мг межа безпеки щодо впливу на ембріофетальний та пре-/постнатальний розвиток відсутня. У годуючих щурів, яким вводили одноразову пероральну дозу 10 мг/кг, сипонімод та його метаболіти проникали у молоко.
Клінічні характеристики.
Показання.
Препарат Кайендра застосовується для лікування дорослих пацієнтів з вторинно-прогресуючим розсіяним склерозом (ВПРС) з активним перебігом, про що свідчить рецидив або ознаки запальної активності.
Протипоказання.
• Підвищена чутливість до діючої речовини препарату – сипонімоду, до арахісу, сої або до будь-якої з допоміжних речовин ( див. розділ «Склад»).
• Синдром імунодефіциту.
• Прогресуюча мультифокальна лейкоенцефалопатія (ПМЛ) або криптококовий менінгіт в анамнезі.
• Злоякісні новоутворення в активній фазі.
• Тяжке порушення функції печінки (клас C за шкалою Чайлда – П’ю) .
• Наявність у пацієнта у період попередніх 6 місяців діагностованих інфаркту міокарда (ІМ), нестабільної стенокардії, інсульту/транзиторної ішемічної атаки (ТІА), декомпенсованої серцевої недостатності, що вимагає стаціонарного лікування, або серцевої недостатності класу III/IV за класифікацією Нью-Йоркської Асоціації Кардіологів (NYHA).
• Наявність в анамнезі пацієнта атріовентрикулярної (АВ) блокади II ступеня Мобітц II, АВ блокади III ступеня, синоатріальної блокади серця або синдрому слабкості синусового вузла, якщо кардіостимулятор не використовується (див. розділ «Особливості застосування»).
• Гомозиготність за генотипом CYP2C9*3 (CYP2C9*3*3).
• Вагітність. Препарат також протипоказаний жінкам репродуктивного віку, які не використовують ефективні методи контрацепції.
Взаємодія з іншими лікарськими засобами та інші види взаємодій.
Фармакокінетичні взаємодії
Імовірність впливу інших лікарських засобів на фармакокінетику сипонімоду
Сипонімод метаболізується головним чином цитохромом P450 2C9 (CYP2C9) (79,3 %) і меншою мірою цитохромом P450 3A4 (CYP3A4) (18,5 %). CYP2C9 є поліморфним ферментом, і генотип впливає на те, наскільки кожен із двох шляхів окисного метаболізму сприяє загальній елімінації. Моделювання PBPK вказує на диференційоване інгібування генотипу CYP2C9 та індукцію сигнальних шляхів CYP3A4. Отже, за прогнозами, ефект міжлікарської взаємодії у присутності речовин, які можуть впливати на CYP3A або CYP2C9, залежить від генотипу CYP2C9 (див. розділи «Особливості застосування» та «Фармакокінетика»).
Генотипи CYP2C9*5, *6, *8 і *11 також пов’язані з частковою або повною втратою активності ферменту CYP2C9. Фармакокінетичні дослідження цих поліморфізмів не проводились. Однак у носіїв цих генотипів спостерігалося підвищення рівня інших субстратів CYP2C9, таких як фенітоїн або варфарин, що потребувало коригування дози цих субстратів (див. також розділи «Особливості застосування» та «Фармакокінетика»).
Інгібітори CYP2C9 та CYP3A4
Одночасне застосування сипонімоду та лікарських засобів, що викликають помірне інгібування CYP2C9 і помірне або потужне інгібування CYP3A4, не рекомендується через значне посилення експозиції сипонімоду. Така схема одночасного застосування лікарських засобів може складатися з помірного подвійного інгібітора CYP2C9/CYP3A4 (наприклад флуконазолу) або помірного інгібітора CYP2C9 в поєднанні з помірним чи потужним інгібітором CYP3A4.
Одночасний прийом флуконазолу (помірного інгібітора CYP2C9/потужного інгібітора CYP3A4) у дозі 200 мг один раз на добу в рівноважному стані і одноразової дози сипонімоду 4 мг у здорових добровольців з генотипом CYP2C9 *1*1 призвів до двократного збільшення площі під кривою (AUC) сипонімоду. Відповідно до оцінки потенціалу лікарської взаємодії із застосуванням фізіологічно обґрунтованого фармакокінетичного (PBPK) моделювання максимальне двократне збільшення AUC сипонімоду прогнозується для генотипів CYP2C9*1*1, *1*2, *1*3 і *2 *3 з будь-яким типом інгібіторів CYP3A4 і CYP2C9. У пацієнтів з генотипом *2*2 гена CYP2C9 очікується збільшення AUC сипонімоду у 2,7 раза в присутності помірних інгібіторів CYP2C9/CYP3A4.
Дані про взаємодію з інгібіторами CYP2C9 та CYP3A4 недоступні для інших генотипів CYP2C9 зі зниженою активністю CYP2C9 або без неї.
Індуктори CYP2C9 та CYP3A4
Через клінічно значуще зниження експозиції сипонімоду потрібна обережність при одночасному застосуванні препарату Кайендра з лікарськими засобами, які спричиняють помірну індукцію CYP2C9 та потужну індукцію CYP3A4. Таке супутнє лікування може включати помірний подвійний індуктор CYP2C9/потужний CYP3A4 (наприклад рифампіцин або карбамазепін) або помірний індуктор CYP2C9 у комбінації з окремим потужним індуктором CYP3A4.
Також необхідна обережність при одночасному застосуванні препарату Кайендра з помірними індукторами CYP3A4 (наприклад модафінілом) або потужними індукторами CYP3A4 пацієнтам із генотипом CYP2C9*1*3 або *2*3, для яких рекомендовано коригування дози (див. розділ «Спеціальні рекомендації щодо дозування»). Дані про взаємодію з індукторами CYP2C9 і CYP3A4 недоступні для інших генотипів CYP2C9 зі зниженою активністю CYP2C9 або без неї.
В цих умовах очікується значуще зниження експозиції сипонімоду (до 76 % і 51 % відповідно) згідно з результатами клінічних досліджень міжлікарської взаємодії та тестів in silico (фізіологічна фармакокінетика) потужних індукторів CYP3A4/ помірних індукторів CYP2C9 (наприклад карбамазепіну) та помірних індукторів CYP3A4 (наприклад модафінілу).
Одночасне застосування 2 мг сипонімоду один раз на добу та 600 мг рифампіцину один раз на добу (потужного індуктора CYP3A4 та помірного індуктора CYP2C9) знижувало AUCtau,ss та Cmax,ss сипонімоду на 57 % та 45 % у пацієнтів з генотипом *1*1 гена CYP2C9.
Сипонімод не є субстратом ефлюксних транспортерів P-gp, BCRP або MRP. Тому передбачається, що лікарські засоби, що впливають на активність цих транспортерів, не впливають на фармакокінетику сипонімоду.
Захоплення сипонімоду в гепатоцитах відбувається виключно шляхом пасивної дифузії. З цієї причини не очікується, що сипонімод взаємодіє з транспортерами печінкового захоплення (OATP, OCT, OAT).
Імовірність впливу сипонімоду на фармакокінетику інших лікарських засобів
Результати дослідження in vitro показали, що сипонімод та його метаболіти (M17 та M3) в терапевтично значущих концентраціях не можуть пригнічувати активність ферментів CYP або можуть пригнічувати тільки незначною мірою (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5), не можуть активувати їх або можуть активувати їх тільки незначною мірою (CYP1A2, CYP2B6, CYP2C9 та CYP3A4/5).
На підставі даних in vitro не очікується, що сипонімод та його метаболіти (M17 та M3) матимуть інгібуючий вплив на захоплення одночасно введених лікарських засобів та/або біологічних активних речовин, що транспортуються OATP1B1, OATP1B3, OAT1, OAT3, OCT1 або OCT2. Очікується також, що вони не будуть інгібувати ефлюкс одночасно введених лікарських засобів та/або біологічних активних речовин, що транспортуються BCRP, BSEP, MATE1, MATE2K або через P-gp в терапевтичних концентраціях.
Фармакодинамічні взаємодії
Протипухлинна, імуномодулююча або імуносупресивна терапія
Застосування препарату Кайендра в комбінації з протипухлинними, імуномодулюючими або імуносупресивними препаратами не вивчалося. Слід дотримуватися обережності при одночасному застосуванні зазначених препаратів із сипонімодом у зв’язку з ризиком розвитку кумулятивного впливу на імунну систему під час терапії, а також протягом декількох тижнів після припинення застосування будь-якого із зазначених препаратів (див. розділ «Особливості застосування»).
При переведенні пацієнтів з іншого препарату, що модифікує захворювання, слід враховувати період напіввиведення і механізм дії іншої терапії, щоб уникнути адитивного імунного ефекту і водночас мінімізувати ризик реактивації захворювання.
Зважаючи на характеристики та тривалість імуносупресуючої дії алемтузумабу, як описано в інформації про фармацевтичний препарат, не рекомендується розпочинати лікування препаратом Кайендра після алемтузумабу, якщо користь від лікування препаратом Кайендра явно не перевищує ризики для окремого пацієнта.
Слід розпочати прийом препарату Кайендра одразу після припинення застосування інтерферону бета або глатирамеру ацетату.
Антиаритмічні засоби, лікарські засоби, що пролонгують інтервал QT, та лікарські засоби, які можуть зменшити частоту серцевих скорочень
На момент початку лікування препаратом Кайендра не слід одночасно застосовувати пацієнтам, які отримують антиаритмічні лікарські засоби класу ІА (наприклад хінідин, прокаїнамід) або класу III (наприклад аміодарон, соталол), лікарські засоби, що пролонгують інтервал QT, з відомими аритмогенними властивостями, блокатори кальцієвих каналів, що знижують ЧСС (такі як верапаміл або дилтіазем), або інші препарати, здатні знижувати ЧСС (наприклад івабрадин або дигоксин), через потенційний адитивний вплив. При розгляді можливості лікування препаратом Кайендра слід отримати консультацію кардіолога до початку терапії (див. розділ «Особливості застосування»).
Бета-адреноблокатори
Слід дотримуватися обережності, починаючи застосування сипонімоду пацієнтам, які отримують бета-адреноблокатори, у зв’язку з можливим розвитком адитивного ефекту зниження ЧСС (див. розділ «Особливості застосування»). Лікування бета-адреноблокаторами може бути розпочато у пацієнтів, які отримують стабільні дози препарату Кайендра.
Негативну хронотропну дію при одночасному застосуванні сипонімоду і пропранололу оцінювали в спеціальному дослідженні фармакодинаміки/безпеки. Додавання пропранололу до терапії сипонімодом на тлі рівноважного фармакокінетичного стану супроводжувалося менш вираженою негативною хронотропною дією (ніж у випадку адитивного впливу) в порівнянні з додаванням сипонімоду до терапії пропранололом в рівноважному фармакокінетичному стані (адитивний вплив на ЧСС).
Вакцинація
Живі атенуйовані вакцини
Оскільки застосування живих атенуйованих вакцин (наприклад вакцини проти вітряної віспи та жовтої лихоманки) може збільшувати ризик розвитку інфекцій, слід уникати імунізації живими атенуйованими вакцинами на тлі терапії препаратом Кайендра і протягом періоду до 4-х тижнів після лікування (див. розділ «Особливості застосування»).
Інші типи вакцин
Під час терапії препаратом, а також протягом періоду до 4-х тижнів після лікування препаратом Кайендра вакцинація може бути менш ефективною. Передбачається, що ефективність вакцинації не знижується при припиненні терапії сипонімодом за 1 тиждень до планованої вакцинації і відновленні прийому препарату не раніше ніж через 4 тижні після її проведення (див. підрозділ «Повторний початок підтримуючої терапії після переривання лікування»). У спеціальному дослідженні фази I за участю здорових добровольців рівень відповіді на квадривалентну вакцину проти грипу, яку вводили після максимум 10 днів попереднього лікування сипонімодом без переривання такого лікування або під час паузи в лікуванні від 10 днів до 14 днів, був приблизно на 15–30 % нижчим порівняно з таким при застосуванні плацебо, тоді як рівень відповіді на вакцинацію PPV 23 при супутньому лікуванні сипонімодом істотно не змінився порівняно з таким при застосуванні плацебо (див. розділ «Особливості застосування»).
Пероральні контрацептиви
Ефективність досліджуваних пероральних контрацептивів (комбінація етинілестрадіолу та левоноргестрелу) підтримувалась під час лікування сипонімодом. Сипонімод не чинив жодного впливу на фармакодинаміку контрацептивів (естрадіолу, прогестерону; ФСГ, ЛГ, розмір фолікулів, оцінка за критеріями Хогланда, ГЗСГ). Порівняно з одноразовим введенням пероральних контрацептивів, одночасне застосування сипонімоду збільшувало площу під кривою левоноргестрелу під час прийому (AUCtau) в 1,29 раза (співвідношення геометричних середніх (СГС): 1,29, 90 % ДІ: 1,24, 1,34) та рівноважні пікові концентрації у плазмі крові (Cmax.ss) у 1,18 раза (СГС: 1,18, 90 % ДІ: 1,11, 1,26). Сипонімод не впливає на фармакокінетику етинілестрадіолу (СГС AUCtau: 1,00, 90 % ДІ: 0,96, 1,05; Cmax,ss GMR: 1,02, 90 %: 0,96, 1,08).
Жодні дослідження взаємодії з пероральними контрацептивами, що містять інші прогестагени, не проводились.
Особливості застосування.
Інфекції
Основним фармакодинамічним ефектом препарату Кайендра є дозозалежне зниження кількості лімфоцитів в периферичній крові до 20–30 % від вихідних значень. Це пов’язано з оборотним перерозподілом лімфоцитів в лімфоїдних тканинах (див. розділ «Фармакологічні властивості»).
Вплив препарату Кайендра на імунну систему може збільшити ризик розвитку інфекцій.
Перед початком лікування препаратом Кайендра потрібно отримати результати останнього повного аналізу крові (тобто протягом останніх 6 місяців або після припинення попередньої терапії). Також через 3-4 місяці після початку лікування також рекомендується оцінювати результати повного аналізу крові, а потім принаймні раз на рік і при перших ознаках інфекції. При підтвердженні абсолютного числа лімфоцитів < 0,2 × 109/л слід знизити дозу до 1 мг, оскільки в клінічних дослідженнях знижували дозу сипонімоду у пацієнтів з абсолютним числом лімфоцитів < 0,2 × 109/л. Підтверджене абсолютне число лімфоцитів < 0,2 × 109/л у пацієнтів, які вже отримують сипонімод в дозі 1 мг, має призвести до тимчасового припинення терапії сипонімодом доти, доки рівень не досягне 0,6 × 109/л, при якому можна буде розглянути можливість повторного початку лікування сипонімодом.
У пацієнтів з тяжкою активною інфекцією початок лікування препаратом Кайендра слід відкласти до одужання. Оскільки залишкові фармакодинамічні ефекти, такі як зниження впливу на число лімфоцитів у периферичній крові, можуть зберігатися протягом 3–4 тижнів після відміни препарату, протягом усього цього періоду слід продовжувати спостереження за перебігом інфекції (див. розділ «Припинення терапії»).
Пацієнтів, які приймають препарат Кайендра, слід проінструктувати щодо негайного повідомлення свого лікаря про симптоми інфекції. Щодо пацієнтів із симптомами інфекції під час терапії повинні застосовуватися ефективні діагностичні та терапевтичні стратегії. Якщо у пацієнта розвивається серйозна інфекція, лікування препаратом Кайендра слід призупинити.
Повідомлялося про випадки криптококового менінгіту (КМ) пов’язані з застосуванням препарату Кайендра. Повідомлялося про випадки КМ при використанні іншого модулятора сфінгозин-1-фосфатних рецепторів (S1P). Лікарі повинні бути пильними щодо клінічних симптомів або ознак КМ. Для пацієнтів із симптомами та ознаками КМ слід провести оперативну діагностичну оцінку. Лікування препаратом Кайендра слід призупинити до виключення КМ. У разі діагностування КМ слід негайно розпочати відповідне лікування. Надалі відновлення терапії препаратом Кайендра у таких випадках протипоказано (див. розділ «Протипоказання»).
Повідомлялося про випадки прогресуючої мультифокальної лейкоенцефалопатії (ПМЛ) для модулятора рецептора S1P, у тому числі препарату Кайендра, та інших методів лікування РС (див. «Побічні реакції»). Лікарі повинні уважно спостерігати за клінічними симптомами або результатами магнітно-резонансної томографії (МРТ), які можуть свідчити про наявність ПМЛ. При підозрі на ПМЛ лікування препаратом Кайендра слід призупинити до виключення ПМЛ. Якщо ПМЛ підтверджено, необхідно негайно розпочати відповідне лікування. Надалі відновлення терапії препаратом Кайендра після підтвердження ПМЛ протипоказано (див. розділ «Протипоказання»).
Були зареєстровані випадки інфікування вірусом герпесу (включаючи випадки менінгіту або менінгоенцефаліту, спричинені вірусом вітряної віспи (VZV)) на тлі застосуванні препарату Кайендра. Пацієнтів, у яких відсутні документально підтверджені дані про перенесену вітряну віспу або про повний курс вакцинації проти VZV, слід обстежити для виявлення антитіл до VZV перед початком терапії препаратом Кайендра (див. підрозділ «Вакцинація»).
Вакцинація
При відсутності антитіл до вірусу VZV пацієнту слід провести повний курс вакцинації до початку терапії препаратом Кайендра, яке слід відкласти на 1 місяць до розвитку повної імунної відповіді на вакцинацію (див. розділ «Побічні реакції»).
Живі атенуйовані вакцини
Оскільки застосування живих атенуйованих вакцин (наприклад, вакцина проти вітряної віспи та вакцина проти жовтої лихоманки) може становити ризик інфікування, його слід уникати під час прийому препарату Кайендра і протягом 4 тижнів після припинення лікування цим препаратом (див. розділ «Взаємодія з іншими лікарськими засобами та інші види взаємодій»).
Інші типи вакцин
Ефективність вакцинації іншими типами вакцин може знижуватися при її виконанні під час лікування препаратом Кайендра. Рекомендується перервати лікування за 1 тиждень до планової вакцинації і на 4 тижні після цього. Рішення щодо продовження або припинення лікування препаратом Кайендра слід приймати на основі оцінки користі та ризику для окремого пацієнта (див. підрозділ «Припинення терапії сипонімодом» та розділ «Взаємодія з іншими лікарськими засобами та інші види взаємодій»).
При припиненні терапії сипонімодом для вакцинації слід враховувати можливе повернення активності захворювання (див. підрозділ «Припинення терапії сипонімодом»).
Супутнє лікування протипухлинними, імуномодулюючими або імуносупресивними препаратами
Протипухлинну, імуномодулюючу або імуносупресивну терапію (включаючи кортикостероїди) слід призначати з обережністю у зв’язку з ризиком адитивного впливу на імунну систему під час такої терапії (див. розділ «Взаємодія з іншими лікарськими засобами та інші види взаємодій»).
Макулярний набряк
Макулярний набряк (див. розділ «Побічні реакції») з візуальними симптомами або без них частіше відзначався при застосуванні сипонімоду (1,8 %), ніж плацебо (0,2 %) в клінічній фазі III дослідження А2304. Більшість випадків виникало протягом перших 3–4 місяців терапії. Тому через 3–4 місяці після початку лікування рекомендується проведення офтальмологічної оцінки. Оскільки випадки макулярного набряку також спостерігалися при довгостроковому лікуванні, пацієнтам слід повідомляти про порушення зору в будь-який час в ході терапії препаратом Кайендра, а також рекомендується провести огляд очного дна, включаючи макулярну ділянку.
Препарат слід застосовувати з обережністю пацієнтам з цукровим діабетом, увеїтом або основним/супутнім захворюванням сітківки у зв’язку з потенційним підвищенням ризику розвитку макулярного набряку. Цим пацієнтам рекомендується проходження офтальмологічного обстеження до початку терапії і регулярно в ході лікування препаратом Кайендра з метою виявлення макулярного набряку.
Продовження терапії препаратом Кайендра у пацієнтів з макулярним набряком не оцінювалось. При розвитку у пацієнта макулярного набряку рекомендується припинити прийом сипонімоду. При прийнятті рішення про те, чи слід повторно починати лікування препаратом Кайендра після зникнення макулярного набряку, необхідно враховувати потенційну користь і ризики для пацієнта в кожному конкретному випадку.
Брадикардія та брадіаритмія
Частота пульсу
Через ризик серйозних порушень серцевого ритму або значної брадикардії препарат Кайендра не слід застосовувати пацієнтам з такими порушеннями:
• наявність в анамнезі зупинки серця більше ніж за 6 місяців до початку лікування препаратом Кайендра;
• цереброваскулярні захворювання;
• симптоматична брадикардія або рецидивна непритомність в анамнезі;
• неконтрольована гіпертензія або
• тяжке неліковане апное уві сні.
Якщо лікування розглядається, перед початком лікування слід отримати консультацію кардіолога для визначення найбільш відповідної стратегії моніторингу.
Для таких пацієнтів лікування сипонімодом слід розглядати тільки у разі, коли очікувана користь перевищує потенційні ризики.
Докладне дослідження QT не продемонструвало значного прямого пролонгованого QT ефекту, і препарат Кайендра не асоціювався із аритмогенним потенціалом, пов’язаним з подовженням інтервалу QT. Початок лікування може призвести до зниження ЧСС і непрямого подовження інтервалу QT під час фази титрування. Не вивчалося застосування препарату Кайендра пацієнтам зі значним подовженням інтервалу QT (QTc більше 500 мсек) або пацієнтам, які отримували лікарські засоби, що пролонгують інтервал QT. Якщо лікування препаратом Кайендра розглядається для пацієнтів з попередньо існуючою клінічно значущою пролонгацією інтервалу QT або для пацієнтів, які вже перебувають на лікуванні лікарськими засобами, що пролонгують QT, з відомими аритмогенними властивостями, слід до початку лікування отримати консультацію кардіолога для визначення найбільш відповідної стратегії моніторингу на початку лікування.
Не вивчалося застосування препарату Кайендра пацієнтам з аритміями, які вимагають застосування антиаритмічних лікарських засобів класу ІА (наприклад, хінідин, прокаїнамід) або класу III (наприклад, аміодарон, соталол). Застосування антиаритмічних лікарських засобів класу ІА та III асоційоване з випадками розвитку шлуночкової тахікардії типу «пірует» у пацієнтів з брадикардією. Оскільки на початку лікування відбувається зниження ЧСС, препарат Кайендра не слід застосовувати одночасно з цими лікарськими засобами на момент початку лікування.
Обмежений досвід застосування препарату пацієнтам, які отримували супутню терапію блокаторами кальцієвих каналів, що знижують ЧСС (такими як верапаміл або дилтіазем), або іншими препаратами, які можуть знижувати ЧСС (наприклад, івабрадин або дигоксин). Не вивчалось застосування цих лікарських засобів пацієнтам, які отримували препарат Кайендра, в клінічних дослідженнях. Одночасний прийом цих препаратів на момент початку лікування може бути пов’язаний з тяжкою брадикардією і серцевої блокадою. Як правило, пацієнтам, які одночасно приймають ці препарати, не слід починати лікування препаратом Кайендра через потенційний адитивний вплив на ЧСС (див. розділ «Взаємодія з іншими лікарськими засобами та інші види взаємодій»).
Якщо на момент початку лікування препаратом Кайендра розглядається супутнє лікування однією з вищевказаних речовин, слід звернутися до кардіолога щодо переходу на лікарський засіб, що не знижує ЧСС, або щодо відповідного моніторингу для ініціації лікування препаратом Кайендра.
Початок лікування препаратом Кайендра призводить до тимчасового зниження ЧСС (див. розділ «Побічні реакції»), і тому на початку лікування застосовується схема титрування дози для досягнення підтримуючої дози препарату Кайендра на шостий день (див. розділ «Спосіб застосування та дози»).
Після першого титрування дози зниження ЧСС починається протягом однієї години, а зниження в перший день стає максимальним приблизно через 3–4 години. При продовженні поступового підвищення дози подальше зниження ЧСС спостерігається в наступні дні, причому максимальне зниження в порівнянні з першим днем (вихідний рівень) досягається на 5–6-й день. Найбільше добове зниження абсолютної середньогодинної ЧСС після прийому дози спостерігається в перший день, при цьому частота пульсу знижується в середньому на 5–6 ударів за хвилину (уд/хв). Зниження після прийому дози в наступні дні менш виражене. При продовженні прийому доз серцевий ритм починає збільшуватися через 6 днів і досягає рівня, що спостерігається при прийомі плацебо, протягом 10 днів після початку лікування.
Значення ЧСС, нижче 40 ударів за хвилину, спостерігалися рідко. У пацієнтів з розвитком брадикардії симптоми були відсутні. У декількох пацієнтів виникали симптоми від легкого до помірного ступеня тяжкості, в тому числі запаморочення і втома, які зникали протягом 24 годин без втручання (див. розділ «Побічні реакції»). У разі необхідності зниження ЧСС, спричинене сипонімодом, можна усунути шляхом прийому парентеральних доз атропіну або ізопреналіну.
Атріовентрикулярна провідність
Початок терапії препаратом Кайендра супроводжувався транзиторним уповільненням атріовентрикулярної провідності, яка має часовий характер, подібний до такого у спостережуваного зниження ЧСС у фазі титрування. Уповільнення атріовентрикулярної провідності проявляється в більшості випадків у вигляді атріовентрикулярної (АВ) блокади I ступеня (подовжений PR-інтервал на електрокардіограмі). У клінічних дослідженнях АВ-блокада II ступеня, зазвичай типу Мобітц I (блокада Венкебаха), спостерігалася у менш ніж у 1,7 % пацієнтів на момент початку лікування препаратом Кайендра. Порушення провідності зазвичай були минущими, безсимптомними, усувалися протягом 24 годин і не вимагали припинення лікування препаратом Кайендра.
Рекомендації щодо ініціювання лікування
Як запобіжний захід протягом 6 годин після прийому першої дози препарату Кайендра повинні перебувати під наглядом щодо наявності ознак і симптомів брадикардії пацієнти з такими серцевими захворюваннями:
• синусова брадикардія (ЧСС нижче < 55 уд/хв);
• АВ-блокада І або ІІ ступеня (типу Мобітц I) в анамнезі;
• інфаркт міокарда в анамнезі або серцева недостатність в анамнезі, за умови відсутності протипоказань.
Для цих пацієнтів рекомендується проведення електрокардіограми (ЕКГ) перед прийомом дози і в кінці періоду спостереження. Якщо після прийому дози виникає брадіаритмія або симптоми, пов’язані з провідністю, або на ЕКГ через 6 годин після прийому дози відзначається новий початок АВ-блокади II ступеня або вище, або скоригований інтервал QTc 500 мсек і більше, слід розпочати відповідне лікування і продовжувати спостереження до зникнення симптомів/ відновлення показників. У разі якщо потрібно фармакологічне лікування, моніторинг слід продовжувати протягом ночі з повторенням 6-годинного моніторингу після другої дози.
Брадіаритмічний ефект більш виражений при застосуванні препарату Кайендра одночасно з бета-адреноблокаторами. У пацієнтів, які отримують бета-адреноблокатори в стабільних дозах, слід попередньо оцінити ЧСС в стані спокою. Застосування препарату Кайендра на тлі хронічного прийому бета-блокаторів можливе, якщо ЧСС у спокої > 50 уд/хв. У разі якщо ЧСС в спокої ≤ 50 уд/хв, слід припинити прийом бета-адреноблокатора до відновлення ЧСС > 50 уд/хв. Поновлення терапії бета-адреноблокатором в такому випадку можливе після закінчення фази титрування і досягнення підтримуючої дози препарату Кайендра (див. розділ «Взаємодія з іншими лікарськими засобами та інші види взаємодій»).
Пропущена доза на час початку лікування та відновлення терапії після припинення лікування
Якщо доза титрування пропущена у будь-який день протягом перших шести днів лікування або якщо чотири або більше послідовних щоденних дози лікування пропущені під час проведення підтримуючої терапії, слід застосовувати аналогічне початкове титрування та рекомендації щодо моніторингу (див. розділ «Спосіб застосування та дози»).
Функція печінки
Перед початком терапії препаратом Кайендра слід отримати результати вимірювання рівня трансаміназ та білірубіну (тобто протягом шести місяців до початку терапії). У дослідженні А2304 у 5,6 % пацієнтів, що отримували препарат Кайендра у дозі 2 мг, та у 1,5 % пацієнтів, що отримували плацебо, відмічалось збільшення активності аланінамінотрансферази (АЛТ) або аспартатамінотрансферази (АСТ), що втричі перевищує верхню межу норми (ВМН) (див. розділ «Побічні реакції»). У клінічних дослідженнях лікування препаратом Кайендра припиняли в разі підвищення ВМН більше ніж у 3 рази і наявності у пацієнта симптомів, пов’язаних з порушенням функції печінки, або в разі підвищення ВМН більше ніж у 5 разів.
У пацієнтів з розвитком симптомів, що вказують на порушення функції печінки, таких як нудота невідомої етіології, блювання, біль у животі, втома, анорексія, висип з еозинофілією або жовтяницею та/або темний колір сечі, під час лікування, слід проводити моніторинг рівнів печінкових ферментів і припинити прийом препарату Кайендра у разі підтвердження значного ураження печінки. Поновлення терапії буде залежати від того, чи буде визначена інша причина ураження печінки, і від користі для пацієнта від відновлення терапії в порівнянні з ризиком рецидиву порушення функції печінки.
Хоча немає жодних даних, які дають змогу встановити, що у пацієнтів з уже існуючим захворюванням печінки при прийомі препарату Кайендра зростає ризик підвищення печінкових проб, слід дотримуватися обережності пацієнтам із клінічно значущим захворюванням печінки в анамнезі.
Шкірні новоутворення
У дослідженні A2304 базальноклітинна карцинома (БКК) була найпоширенішим новоутворенням, про яке зі схожою частотою повідомлялася в групах застосування сипонімоду 2 мг (1,1 %, 12 пацієнтів) і плацебо (1,3 %, 7 пацієнтів). Частота плоскоклітинної карциноми (ПКК) у дослідженні A2304 була однаковою (0,2 %) у пацієнтів, які отримували препарат Кайендра, і пацієнтів, які отримували плацебо. У довгостроковому дослідженні спостерігалося невелике збільшення частоти виникнення BCC та SCC під час тривалого застосування.
Проте у пацієнтів, які отримували сипонімод, і у пацієнтів, які отримували тривалу терапію іншим модулятором S1P, також були зареєстровані інші злоякісні новоутворення шкіри, включаючи меланому.
Огляд шкіри рекомендується проводити всім пацієнтам на початку лікування, а потім кожні 6–12 місяців, зважаючи на клінічну оцінку.
Пацієнтам слід рекомендувати негайно повідомляти свого лікаря про будь-які підозрілі ураження шкіри.
Пацієнтів, які отримують лікування сипонімодом, слід попереджати про необхідність застосування засобів захисту від впливу сонячного світла. Крім того, показані регулярні огляди у дерматолога, особливо пацієнтам з відомими факторами ризику розвитку пухлин шкіри та пацієнтам з відомими підозрілими ураженнями шкіри. Пацієнтам, які приймають препарат Кайендра, не слід отримувати супутню фототерапію з ультрафіолетовим випромінюванням типу В або ПУВА-фотохіміотерапію.
Непередбачені неврологічні або психічні симптоми/ознаки
Рідкісні випадки розвитку синдрому задньої оборотної енцефалопатії (PRES) були зареєстровані при застосуванні іншого модулятора сфінгозин-1-фосфатних рецепторів (S1P). Такі події не відмічались при застосуванні препарату Кайендра в рамках програми клінічних досліджень. Однак у разі розвитку будь-яких непередбачених неврологічних або психічних симптомів чи ознак (наприклад, когнітивний розлад, зміна поведінки, кортикальні зорові порушення або будь-які інші кортикальні неврологічні симптоми/явища чи будь-які інші симптоми/явища, що дають змогу запідозрити підвищення внутрішньочерепного тиску) або при стрімкому погіршенні неврологічного статусу на тлі терапії препаратом слід негайно провести повну оцінку фізичного і неврологічного статусу і розглянути можливість проведення МРТ.
Попередня терапія імуносупресорами або імуномодуляторами
При переведенні пацієнтів з іншого препарату, що модифікує захворювання, слід враховувати період напіввиведення і механізм дії іншої терапії, щоб уникнути адитивного імунного ефекту і водночас мінімізувати ризик реактивації захворювання. Перед початком лікування препаратом Кайендра рекомендується визначити число лімфоцитів в периферичній крові (CBC), щоб переконатися, що вплив попередньої терапії на імунну систему (тобто цитопенія) вже усунений.
Вплив на артеріальний тиск
Пацієнти з артеріальною гіпертензією, що не контролюється лікарськими препаратами, не допускалися до участі в передреєстраційних клінічних дослідженнях, і при лікуванні сипонімодом таких пацієнтів з неконтрольованою гіпертензією необхідно проявляти особливу обережність.
У дослідженні А2304 за участю пацієнтів з ВПРС артеріальна гіпертензія частіше відзначалася у пацієнтів, що приймали сипонімод (12,6 %), ніж у пацієнтів, які отримували плацебо (9,0 %). Лікування сипонімодом призводило до підвищення систолічного і діастолічного артеріального тиску, яке розпочиналося рано після початку лікування, досягало максимуму приблизно через 6 місяців (систолічний тиск – 3 мм рт. ст., діастолічний тиск – 1,2 мм рт. ст.) і залишалося стабільним пізніше. Цей ефект зберігається при продовженні лікування.
Під час застосування сипонімоду та лікування артеріальної гіпертензії необхідно проводити регулярний моніторинг артеріального тиску.
Генотип CYP2C9
Перед початком лікування препаратом Кайендра необхідно провести генотипування пацієнтів на CYP2C9 для визначення їхнього статусу метаболізатора CYP2C9 (див. розділи «Протипоказання», «Спосіб застосування та дози» та «Фармакокінетика»). Пацієнтам, які є гомозиготними носіями генотипу CYP2C9*3 (*3*3 гена CYP2C9) (приблизно від 0,3 до 0,4 % населення, що представляє народи Кавказу; рідше – інші етноси), не слід призначати лікування препаратом Кайендра, оскільки застосування препарату Кайендра у цих пацієнтів призводить до суттєвого підвищення рівнів сипонімоду в плазмі крові (див. розділи «Фармакокінетика» та «Протипоказання»).
Рекомендована підтримуюча доза препарату Кайендра для носіїв генотипу *2*3 гена CYP2C9 (1,4–1,7 % пацієнтів) та для пацієнтів з генотипом *1*3 (9–12 % пацієнтів) становить 1 мг на добу, щоб уникнути підвищеної експозиції сипонімоду (див. розділи «Спосіб застосування та дози» та «Фармакокінетика»).
Вплив генотипів, відмінних від *2 і *3, на фармакокінетику сипонімоду не вивчався. Незважаючи на те, що дослідження впливу більш рідкісних алелів CYP2C9*5, *6, *8 і *11 на метаболізм сипонімоду не проводилися, підвищення рівня сипонімоду не можна виключити через зниження або втрату активності ферменту у носіїв цих поліморфізмів CYP2C9 (див. також розділи «Взаємодія з іншими лікарськими засобами та інші види взаємодій» та «Фармакокінетика»). Загальна частота 4 алелів *5, *6, *8 і *11 становить 10 % у африканців/осіб африканського походження, 2 % у латиноамериканців і < 0,4% у кавказців та азіатів. Однак через недостатню кількість даних для цих генотипів неможливо дати рекомендації щодо будь-якої корекції дози.
У ході клінічних досліджень при застосуванні нескоригованої добової дози 2 мг сипонімоду у гетерозиготних носіїв алелів CYP2C9 *2 і *3, які характеризуються зниженим метаболізмом CYP2C9, не спостерігалося жодних специфічних значущих клінічних симптомів гострої токсичності. Спостерігалося незначне збільшення частоти макулярного набряку після тривалого впливу (див. також розділи «Взаємодія з іншими лікарськими засобами та інші види взаємодій» та «Фармакокінетика»). Однак незрозуміло, чи пов’язані ці відмінності з підвищеним впливом сипонімоду.
Жінки репродуктивного віку
Через ризик для плода застосування сипонімоду протипоказано вагітним та жінкам репродуктивного віку, які не використовують ефективні засоби контрацепції. Перед початком лікування жінок репродуктивного віку необхідно поінформувати щодо цього ризику для плода; вони повинні отримати негативний результат тесту на вагітність і використовувати ефективні засоби контрацепції під час лікування і щонайменше протягом 10 днів після припинення лікування (див. розділи «Протипоказання», «Застосування у період вагітності або годування груддю»).
Припинення терапії сипонімодом
У рідкісних випадках після припинення застосування іншого модулятора рецептора S1P повідомлялося про тяжке загострення захворювання, включаючи відновлення його симптомів. Слід розглянути можливість серйозного загострення захворювання після припинення лікування сипонімодом. Пацієнти повинні перебувати під наглядом для виявлення відповідних ознак можливого серйозного загострення або повернення високої активності захворювання після припинення прийому сипонімоду і для призначення відповідного лікування по мірі необхідності.
Після припинення терапії препарат Кайендра залишається в крові до 10 днів. Початок інших видів терапії протягом цього періоду призведе до супутнього впливу сипонімоду.
У переважної більшості (90 %) пацієнтів з ВПРС число лімфоцитів повертається до нормального рівня протягом 10 днів після припинення терапії. Однак протягом 3–4 тижнів після прийому останньої дози препарату можуть спостерігатися залишкові фармакодинамічні ефекти, такі як зменшення кількості лімфоцитів в периферичній крові. Застосування імуносупресантів протягом цього періоду може призвести до адитивного впливу на імунну систему, і тому слід дотримуватися обережності протягом 3–4 тижнів після прийому останньої дози.
Вплив на результати гематологічного тестування
Оскільки сипонімод знижує число лімфоцитів в крові шляхом перерозподілу у вторинних лімфоїдних органах, підрахунок лімфоцитів в периферичній крові у пацієнта, який одержував сипонімод, не можна використовувати для оцінки стану субпопуляції лімфоцитів. Для проведення лабораторних тестів з використанням циркулюючих мононуклеарних клітин потрібні більші об’єми крові через зменшення кількості циркулюючих лімфоцитів.
Інші компоненти
Таблетки містять фосфоліпіди з соєвих бобів. Пацієнтам з гіперчутливістю до арахісу або сої не слід приймати цей лікарський засіб (див. розділ «Протипоказання»).
Таблетки містять лактозу. Пацієнтам з рідкісними спадковими проблемами непереносимості галактози, дефіцитом лактази або синдромом глюкозо-галактозної мальабсорбції не слід приймати цей лікарський засіб.
Застосування у період вагітності або годування груддю.
Жінки репродуктивного віку/ контрацепція у жінок
Препарат Кайендра протипоказаний жінкам репродуктивного віку, які не використовують ефективні методи контрацепції (див. розділ «Протипоказання»).
Жінкам репродуктивного віку слід повідомити, що результати досліджень на тваринах показали, шкідливість сипонімоду для плода, що розвивається (див. розділ «Дані доклінічних досліджень»). Жінкам репродуктивного віку необхідно отримати негативний результат тесту на вагітність на початку лікування сипонімодом. Жінки мають використовувати ефективні методи контрацепції (методи, при яких відсоток вагітностей становить менше 1 %) під час лікування препаратом Кайендра і щонайменше протягом 10 днів після припинення лікування (див. розділ «Особливості застосування»).
У разі припинення терапії сипонімодом для планування вагітності слід враховувати можливе повернення активності захворювання.
Вагітність
Немає відповідних даних щодо застосування препарату Кайендра вагітним жінкам для інформування про асоційований з лікарським засобом ризик несприятливих наслідків для розвитку плода. Дослідження на тваринах продемонстрували спричинену сипонімодом ембріотоксичність і фетотоксичність у щурів і кролів, а також тератогенность у щурів, включаючи загибель ембріона і плода та вади розвитку скелета або внутрішніх органів при рівнях експозиції, порівнянних з експозицією добової дози 2 мг у людини (див. розділ «Дані доклінічних досліджень»). Крім того, клінічний досвід застосування іншого модулятора сфінгозин-1-фосфатних рецепторів продемонстрував вдвічі більший ризик виникнення тяжких вроджених вад розвитку при прийомі препарату під час вагітності в порівнянні з частотою, що спостерігається в загальній популяції.
Отже, сипонімод протипоказаний жінкам під час вагітності (див. розділ «Протипоказання»). Прийом сипонімоду слід припинити щонайменше за десять днів до планування вагітності (див. розділ «Особливості застосування»). Якщо під час лікування у жінки настає вагітність, прийом сипонімоду необхідно припинити. Слід надати медичну консультацію щодо ризику шкідливого впливу на плід, пов’язаного з лікуванням, і провести УЗД.
Годування груддю
Невідомо, чи виділяється сипонімод або його метаболіти у грудне молоко людини. Сипонімод і його метаболіти екскретуються у молоко щурів. Не слід застосовувати сипонімод в період грудного вигодовування.
Фертильність
Оцінка впливу сипонімоду на фертильність людини не проводилась. Сипонімод не впливав на репродуктивні органи самців щурів і мавп або на показники фертильності у щурів.
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами.
Сипонімод не чинить або чинить незначний вплив на здатність керувати автомобілем та іншими механізмами.
Однак на початку терапії сипонімодом іноді може виникати запаморочення та брадіаритмія. Таким чином, пацієнтам не слід керувати транспортними засобами та працювати з іншими механізмами протягом першого дня початку лікування сипонімодом (див. розділ «Особливості застосування»).
Спосіб застосування та дози.
Лікування препаратом Кайендра слід розпочинати та проводити під наглядом невропатолога, який має досвід лікування пацієнтів з РС.
Перед початком лікування пацієнтам необхідно провести генотипування на CYP2C9.
Препарат Кайендра не слід застосовувати пацієнтам з генотипом *3*3 гена CYP2C9 (див. також розділи «Протипоказання», «Особливості застосування» та «Фармакокінетика»).
Для пацієнтів з генотипом *2*3 або генотипом *1*3 гена CYP2C9 рекомендована підтримуюча доза становить 1 мг один раз на добу (чотири таблетки по 0,25 мг) (див. також розділи «Особливості застосування» та «Фармакокінетика»).
Через брак даних не можна надати рекомендації щодо дозування для носіїв інших рідкісних алелів CYP2C9, для яких передбачається зниження або відсутність активності CYP2C9, таких як, зокрема, алелі CYP2C9 *5, *6, *8 і *11 (див. також розділи «Протипоказання», «Особливості застосування», «Взаємодія з іншими лікарськими засобами та інші види взаємодій» та «Фармакокінетика»).
Рекомендована підтримуюча доза препарату Кайендра для всіх інших пацієнтів з генотипом CYP2C9 становить 2 мг.
Препарат Кайендра слід приймати один раз на добу, незалежно від вживання їжі. Таблетки, вкриті плівковою оболонкою, Кайендра слід ковтати цілими, запиваючи водою.
Початок терапії
Лікування слід розпочинати з поступового збільшення дози протягом 5 днів.
Стан пацієнтів з певними вже наявними серцевими розладами необхідно контролювати щодо появи ознак та симптомів брадикардії протягом перших 6 годин після прийому першої дози препарату Кайендра (див. розділ «Особливості застосування»).
Лікування починають з прийому дози 0,25 мг один раз на добу в перший і на другий день з подальшим прийомом доз 0,5 мг один раз на добу на третій день, 0,75 мг один раз на добу на четвертий день і 1,25 мг один раз на добу на п’ятий день для досягнення запропонованої пацієнтові підтримуючої дози препарату Кайендра, починаючи з шостого дня (див. таблицю 2).
Протягом перших 6 днів початку лікування рекомендовану добову дозу слід приймати один раз на добу вранці, незалежно від прийому їжі.
Таблиця 2
Режим титрування дози для досягнення підтримуючої дози препарату Кайендра
|
День титрування |
Доза титрування |
Режим титрування |
Упаковка |
|
День 1 |
0,25 мг |
1 × 0,25 мг |
|
|
День 2 |
0,25 мг |
1 × 0,25 мг |
|
|
День 3 |
0,5 мг |
2 × 0,25 мг |
СТАРТОВА УПАКОВКА |
|
День 4 |
0,75 мг |
3 × 0,25 мг |
|
|
День 5 |
1.25 мг |
5 × 0,25 мг |
|
|
День 6 |
2 мг# |
1 × 2 мг# |
ПІДТРИМУЮЧА ТЕРАПІЯ для генотипів CYP2C9 *1*1,*1*2 або *2*2 гена |
# Рекомендована підтримуюча доза становить 1 мг (4 × 0,25 мг) один раз на добу для пацієнтів з генотипом *2*3 або *1*3 гена CYP2C9. Додаткова доза 0,25 мг на п’ятий день не ставить під загрозу безпеку пацієнта.
Пропущена доза на початку лікування
Якщо доза титрування пропущена у будь-який день протягом перших шести днів лікування, лікування необхідно почати повторно із застосуванням нової упаковки для титрування дози.
Пропущена доза після шостого дня
У разі пропуску дози подальшу дозу слід приймати у звичний час на наступний день; цю наступну дозу не потрібно подвоювати.
Повторний початок підтримуючої терапії після переривання лікування
У разі переривання підтримуючої терапії препаратом Кайендра на чотири або більше послідовних щоденних дози, прийом препарату необхідно розпочати повторно із застосуванням нової упаковки для титрування дози (див. розділ «Початок лікування»). У разі переривання терапії до 4 пропущених послідовних щоденних доз повторне титрування не потрібне, а терапія повинна проводитися підтримуючою дозою.
Спеціальні рекомендації щодо дозування
Пацієнти з порушенням функції печінки
Сипонімод не слід приймати пацієнтам з тяжким порушенням функції печінки (клас C за шкалою Чайлда – П’ю) (див. розділ «Протипоказання»). Незважаючи на те, що корекція дози не потрібна пацієнтам з легким або помірним порушенням функції печінки, слід з обережністю розпочинати лікування цій категорії пацієнтів (див. розділи «Особливості застосування», «Фармакокінетика»).
Пацієнти з порушенням функції нирок
Корекція дози препарату Кайендра не потрібна пацієнтам з порушенням функції нирок.
Пацієнти літнього віку
Не вивчалося застосування препарату Кайендра пацієнтам віком понад 65 років. У клінічних дослідженнях брали участь пацієнти віком до 61 року. Препарат Кайендра слід з обережністю застосовувати пацієнтам літнього віку у зв’язку з недостатністю клінічних даних щодо ефективності та безпеки препарату.
Діти.
Дослідження за участю дітей не проводились.
Передозування.
Пацієнти отримували сипонімод у вигляді одноразових доз (від 0,1 до 75 мг) або у вигляді багаторазових доз (від 0,25 до 20 мг). На підставі виникнення симптоматичної брадикардії після одноразового прийому 75 мг було визначено, що для здорових осіб одноразова максимальна переносима доза становить 25 мг. Найвища досліджувана багаторазова доза 20 мг протягом 28 днів переносилась добре (9 пацієнтів отримували 100 мг в останній день дозування, а 5 пацієнтів випадково отримували до 200 мг один раз на добу протягом 3–4 днів). Деякі з 9 досліджуваних відмічали безсимптомне легке та помірне підвищення печінкових проб.
Один пацієнт (з депресією в анамнезі) приймав 84 мг сипонімоду. У цього пацієнта відмічалось незначне підвищення рівня печінкових трансаміназ. У пацієнта не відмічалось жодних інших побічних реакцій після передозування.
У разі передозування при першому прийомі препарату або під час фази титрування слід контролювати стан пацієнта з метою виявлення можливих ознак і симптомів брадикардії, можливе продовження спостереження до ранку наступного дня. Необхідно регулярно контролювати артеріальний тиск, частоту пульсу та провести ЕКГ (див. розділи «Спосіб застосування та дози» та «Особливості застосування»).
Специфічного антидоту для сипонімоду не існує. Сипонімод не видаляється в значній кількості з організму шляхом гемодіалізу або плазмаферезу.
Побічні реакції.
Під час клінічного дослідження ІІІ фази A2304 1651 пацієнт з ВПРС був рандомізований у співвідношенні 2:1 для отримання препарату Кайендра в дозі 2 мг один раз на добу або плацебо. Медіана тривалості лікування становила 18 місяців (від 0 до 37 місяців). На момент реєстрації препарату довгострокові дані про безпеку дуже обмежені. Найпоширенішими побічними реакціями на тлі прийому 2 мг сипонімоду є головний біль (15,2 %) та гіпертензія (12,6 %).
Побічні реакції, що спостерігались у клінічних дослідженнях, були визначені насамперед на підставі досвіду застосування в рамках основного дослідження A2304 (таблиця 3) та наведені нижче згідно з класами систем органів за класифікацією MedDRA.
В межах кожного класу систем органів побічні реакції розподілені за частотою, при цьому найчастіші вказуються першими. Крім того, відповідні категорії частоти для кожної небажаної реакції на препарат вказано з використанням таких умовних позначень (CIOMS ІІІ): дуже часто (≥ 1/10); часто (від ≥ 1/100 до < 1/10); нечасто (від ≥ 1/1000 до < 1/100); рідко (від ≥ 1/10000 до < 1/1000); дуже рідко (< 1/10000), невідомо (неможливо оцінити за наявними даними).
Таблиця 3
Список побічних реакцій у вигляді таблиці
|
Інфекції та інвазії |
||
|
Часто |
Оперізуючий герпес |
|
|
Нечасто |
Криптококовий менінгіт *# |
|
|
Рідко |
Прогресуюча мультифокальна лейкоенцефалопатія *# |
|
|
Доброякісні, злоякісні та неуточнені новоутворення (включаючи кісти та поліпи) |
||
|
Часто |
Меланоцитарний невус # Базальноклітинна карцинома *# |
|
|
Нечасто |
Плоскоклітинна карцинома *# |
|
|
Порушення з боку крові та лімфатичної системи |
||
|
Часто |
Лімфопенія |
|
|
Порушення з боку нервової системи |
||
|
Дуже часто |
Головний біль |
|
|
Часто |
Запаморочення Судоми Тремор |
|
|
Порушення з боку органів зору |
||
|
Часто |
Макулярний набряк |
|
|
Порушення з боку серця |
||
|
Часто |
Брадикардія Атріовентрикулярна блокада (першого та другого ступеня) |
|
|
Порушення з боку судин |
||
|
Дуже часто |
Артеріальна гіпертензія # |
|
|
Порушення з боку шлунково-кишкового тракту |
||
|
Часто |
Нудота Діарея |
|
|
Порушення з боку скелетно-м’язової системи і сполучної тканини |
||
|
Часто |
Біль в кінцівках |
|
|
Загальні розлади та реакції в місці застосування |
||
|
Часто |
Периферичний набряк Астенія |
|
|
Лабораторні дані |
||
|
Дуже часто |
Підвищення показників печінкових проб |
|
|
Часто |
Зниження показників функції легень |
|
# Дивіться також розділ «Особливості застосування».
* Побічні реакції на лікарські засоби, виявлені у відкритому продовженні фази 3 дослідження A2304.
Опис окремих побічних реакцій
Інфекції
Під час клінічного дослідження А2304 загальна частота інфекцій у пацієнтів з вторинно-прогресуючим розсіяним склерозом (ВПРС) була порівнянна у пацієнтів, що приймали сипонімод, та у тих, хто отримував плацебо (49,0 % і 49,1 % відповідно). Однак повідомлялося про збільшення частоти оперізувального герпесу у пацієнтів, що приймали сипонімод (2,5 %), в порівнянні з пацієнтами, що приймали плацебо (0,7 %). Не спостерігалося подальшого підвищення рівня інфекційних захворювань, спричинених вірусом вітряної віспи, при тривалому впливі сипонімоду. Також повідомлялося про випадки менінгіту або менінгоенцефаліту, спричинених вірусом вітряної віспи, під час терапії препаратом Кайендра (див. розділ «Особливості застосування»).
Повідомлялося про випадки прогресуючої мультифокальної лейкоенцефалопатії та криптококового менінгіту (КМ) на тлі застосування препарату Кайендра (див. розділи «Особливості застосування» та «Протипоказання»).
Макулярний набряк
Макулярний набряк частіше відзначався у пацієнтів, які приймали сипонімод (1,8 %), ніж у пацієнтів, які приймали плацебо (0,2 %). Хоча більшість випадків виникала протягом 3–4 місяців після початку прийому сипонімоду, макулярний набряк також був зареєстрований у пацієнтів, які отримували сипонімод від шести до дванадцяти місяців (див. розділ «Особливості застосування»). У деяких пацієнтів відзначалася нечіткість зору або зниження гостроти зору, проте у інших хвороба протікала безсимптомно і порушення було діагностовано при плановому офтальмологічному обстеженні. Загалом після припинення прийому препарату відзначалося покращення або спонтанне зникнення макулярного набряку. Оцінку ризику розвитку рецидиву після відновлення прийому препарату не проводили.
Брадикардія
На початку терапії сипонімодом може відзначатися тимчасове зниження ЧСС, а також уповільнення атріовентрикулярної провідності (див. розділ «Особливості застосування»). Брадикардія повідомлялася у 6,2 % пацієнтів, які отримували сипонімод, в порівнянні з 3,1 % пацієнтів, які отримували плацебо, а АВ-блокада відзначалася у 1,7 % пацієнтів, які отримували сипонімод, в порівнянні з 0,7 % пацієнтів, які отримували плацебо.
Максимальне зниження ЧСС спостерігається в перші 6 годин після прийому дози.
Тимчасове дозозалежне зниження ЧСС спостерігалося під час початкової фази дозування і досягало плато при дозах 5 мг і більше. Брадіаритмічні явища (АВ-блокада і синусові паузи) спостерігалися з більшою частотою при лікуванні сипонімодом в порівнянні з плацебо.
Більшість випадків АВ-блокади і синусових пауз виникали при терапевтичній дозі 2 мг, причому частота виникнення в умовах нетитрування дози значно вища, ніж в умовах титрування.
Зниження ЧСС, спричинене сипонімодом, можна усунути атропіном або ізопреналіном.
Печінкові проби
Повідомлялося про підвищення рівнів печінкових ферментів (в основному про підвищення рівня АЛТ) у пацієнтів з РС, які приймали сипонімод.
Під час клінічного дослідження А2304 за участю пацієнтів з ВПРС підвищення печінкових проб частіше спостерігалось у пацієнтів, які приймали сипонімод (11,3 %), ніж у пацієнтів, які приймали плацебо (3,1 %), переважно через підвищення рівнів печінкових трансаміназ (АЛТ/АСТ/ГГТ). Значна частина випадків підвищення печінкових проб зареєстрована протягом 6 місяців після початку терапії. Повернення показників активності АЛТ до норми відмічалося приблизно протягом 1 місяця після припинення прийому сипонімоду (див. розділ «Особливості застосування»).
Артеріальний тиск
У клінічному дослідженні III фази за участю пацієнтів з ВПРС артеріальна гіпертензія частіше відзначалася у пацієнтів, які приймали сипонімод (12,6 %), ніж у пацієнтів, які отримували плацебо (9,0 %) (див. розділ «Особливості застосування»).
Судоми
У клінічному дослідженні А2304 за участю пацієнтів з ВПРС судоми були зареєстровані у 1,7 % пацієнтів, які отримували сипонімод, в порівнянні з 0,4 % пацієнтів, які приймали плацебо. Невідомо, чи були ці події пов’язані з наслідками РС, з прийомом сипонімоду або цими двома явищами одночасно.
Вплив на дихальну систему
При лікуванні сипонімодом спостерігалося незначне зменшення об’єму форсованого видиху за 1 секунду (ОФВ1) і дифузійної здатності легень для монооксиду вуглецю (DLCO). Через три і шість місяців лікування в клінічному дослідженні А2304 середні зміни щодо вихідного рівня ОФВ1 у пацієнтів з ВПРС в групі сипонімоду становили 0,1 л в кожній часовій точці при відсутності змін в групі плацебо. Ці результати були дещо вищі (середня зміна приблизно на 0,15 л щодо вихідного рівня ОФВ1) у пацієнтів з порушеннями з боку дихальної системи, такими як хронічне обструктивне захворювання легень (ХОЗЛ) або астма, які отримували сипонімод. При тривалому лікуванні таке зниження не призводило до клінічно значущих небажаних явищ і не було пов’язане зі збільшенням числа повідомлень про кашель або задишку.
Повідомлення про побічні реакції після реєстрації лікарського засобу має важливе значення. Це дає змогу проводити моніторинг співвідношення користь/ризик при застосуванні цього лікарського засобу. Медичним та фармацевтичним працівникам, а також пацієнтам або їхнім законним представникам слід повідомляти про усі випадки підозрюваних побічних реакцій та відсутності ефективності лікарського засобу через Автоматизовану інформаційну систему з фармаконагляду за посиланням: https://aisf.dec.gov.ua.
Термін придатності.
18 місяців.
Умови зберігання.
Зберігати у холодильнику (2–8 °C). Зберігати в оригінальній упаковці.
Зберігати у недоступному для дітей місці.
Упаковка.
Для дозування 0,25 мг:
по 12 таблеток у блістері; по 1 блістеру в пеналі; по 1 пеналу у картонній коробці.
Для дозування 2 мг:
по 14 таблеток у блістері; по 2 блістери у картонній коробці.
Категорія відпуску.
За рецептом.
Виробник.
1. Новартіс Фарма ГмбХ / Novartis Pharma GmbH.
2. Новартіс Фармасьютика, С.А. / Novartis Farmaceutica, S.A.
Місцезнаходження виробника та адреса місця провадження його діяльності.
1. Рунштрассе 25, Гостенхоф, Нюрнберг, Баварія, 90429, Німеччина / Roonstrasse 25, Gostenhof, Nuremberg, Bavaria, 90429, Germany.
2. Гран Віа де лес Кортс Каталанес 764, Барселона, 08013, Іспанія / Gran Via de les Corts Catalanes 764, Barcelona, 08013, Spain.