
АМЕЛІВУ
-
Республіка Корея САМСУНГ БІОЕПІС КО., ЛТД.Dosage form: розчин для ін'єкцій, 10 мг/мл у флаконі з боросилікатного скла типу І з гумовою пробкою та алюмінієвим ковпачком з кришкою flip-off; по 1 флакону в стандартно-експортній упаковці в картонній коробціComposition: 1 флакон містить 2,3 мг ранібізумабу в 0,23 мл розчину (10 мг/мл)Medicinal dispensing terms: за рецептомСertificate number: UA/20540/01/01ATS code: S01LA04Expiry: 30 місяців
ІНСТРУКЦІЯ
для медичного застосування лікарського засобу
АМЕЛІВУ
(AMELIVU)
Склад:
діюча речовина: ранібізумаб;
1 флакон містить 2,3 мг ранібізумабу в 0,23 мл розчину (10 мг/мл);
допоміжні речовини: α,α-трегалози дигідрат; гістидину гідрохлорид, моногідрат; гістидин;
полісорбат 20; вода для ін’єкцій.
Лікарська форма. Розчин для ін’єкцій.
Основні фізико-хімічні властивості: прозорий водний розчин від безбарвного до блідо-жовтого кольору.
Фармакотерапевтична група. Засоби, що застосовуються при судинних захворюваннях очей. Антинеоваскуляризаційні засоби. Ранібізумаб.
Код АТХ S01L A04.
Фармакологічні властивості.
Фармакодинаміка.
Механізм дії
Ранібізумаб являє собою фрагмент рекомбінантного гуманізованого моноклонального антитіла проти людського судинного ендотеліального фактора росту А (VEGF-A). Він має високу спорідненість з ізоформами VEGF-A (наприклад, VEGF110, VEGF121 та VEGF165) і, таким чином, запобігає зв’язуванню VEGF-A із його рецепторами VEGFR-1 та VEGFR-2. Зв’язування VEGF-A із його рецепторами призводить до проліферації ендотеліальних клітин та неоваскуляризації, а також до надмірної проникності судин, що, як вважається, сприяє розвитку неоваскулярної форми вікової макулярної дегенерації (ВМД), патологічної міопії (ПМ) та хоріоїдальної неоваскуляризації (ХНВ) або порушення зору, спричиненого діабетичним макулярним набряком (ДМН) або макулярним набряком, що виник внаслідок тромбозу вен сітківки (ТВС).
Клінічна ефективність та безпека
Лікування ексудативної ВМД
Клінічну безпеку та ефективність застосування ранібізумабу вивчали у ході трьох рандомізованих, подвійно маскованих, плацебо- або активно контрольованих досліджень за участю пацієнтів із неоваскулярною ВМД тривалістю 24 місяці. Загалом у цих дослідженнях брали участь 1323 пацієнти (879 активної та 444 контрольної груп).
У дослідженні FVF2598g (MARINA) 716 пацієнтів із мінімальними проявами класичної або прихованої некласичної форми захворювання було рандомізовано у співвідношенні 1:1:1 для введення 1 раз на місяць ранібізумабу в дозі 0,3 мг, ранібізумабу в дозі 0,5 мг або плацебо.
У дослідженні FVF2587g (ANCHOR) 423 пацієнти з переважно класичною формою ХНВ були рандомізовані у співвідношенні 1:1:1 для введення 1 раз на місяць ранібізумабу в дозі 0,3 мг, ранібізумабу в дозі 0,5 мг або фотодинамічної терапії вертепорфіном (вФДТ) (на початку дослідження та кожні 3 місяці потому, якщо флуоресцентна ангіографія свідчила про зберігання чи рецидив проникності судин).
Результати обох досліджень свідчать, що продовження лікування ранібізумабом може також покращити стан у тих пацієнтів, які втратили ≥ 15 літер максимально скоригованої гостроти зору (МСГЗ) протягом першого року лікування.
Статистично значуще покращення зорових функцій, які відзначали пацієнти, згідно з опитуванням Національного офтальмологічного інституту (NEI VFQ-25) спостерігалося на фоні лікування ранібізумабом порівняно з контрольною групою як у дослідженні MARINA, так і у дослідженні ANCHOR.
У дослідженні FVF3192g (PIER) 184 пацієнти з усіма формами неоваскулярної ВМД були рандомізовані у співвідношенні 1:1:1 для введення 1 раз на місяць ранібізумабу в дозі 0,3 мг, ранібізумабу в дозі 0,5 мг або плацебо протягом 3-х послідовних місяців із подальшим введенням дози 1 раз кожні 3 місяці. Починаючи з 14-го місяця дослідження, пацієнти, які отримували плацебо, могли переходити на лікування ранібізумабом, а починаючи з 19-го місяця дозволялося більш часте введення препарату. Пацієнти, які отримували лікування ранібізумабом в дослідженні PIER, отримали в середньому по 10 ін’єкцій препарату.
Після початкового покращення гостроти зору (після введення 1 раз на місяць) в середньому пацієнти, які отримували ранібізумаб 1 раз кожні 3 місяці, втрачали гостроту зору, яка поверталася до початкового рівня на 12-й місяць. Цей ефект зберігався в більшості пацієнтів, які отримували ранібізумаб (82 %), до 24-го місяця. Обмежені дані, отримані у пацієнтів групи плацебо, яким пізніше вводили ранібізумаб, свідчать про те, що ранній початок лікування може забезпечувати краще збереження гостроти зору.
За результатами двох післяреєстраційних досліджень (MONT BLANC, BPD952A2308 та DENALI, BPD952A2309) ефективність ранібізумабу була підтверджена, але вони не продемонстрували додаткової переваги комбінованого застосування вертепорфіну (ФДТ візудином) і ранібізумабу порівняно з монотерапією ранібізумабом.
Лікування порушення зору внаслідок ХНВ, що виникла внаслідок патологічної міопії (ПМ)
Клінічну безпеку та ефективність застосування ранібізумабу в пацієнтів із порушенням зору внаслідок ХНВ при ПМ оцінювали на підставі даних, отриманих протягом 12 місяців у ході проведення рандомізованого, подвійно маскованого, контрольованого базового дослідження F2301 (RADIANCE). У цьому дослідженні 277 пацієнтів були рандомізовані у співвідношенні 2:2:1 у такі групи:
- Група I (ранібізумаб у дозі 0,5 мг, режим дозування встановлювався за критеріями «стабільність зору», які визначалися як відсутність змін максимально скоригованої гостроти зору (МСГЗ) порівняно з результатами двох попередніх щомісячних оцінок).
- Група II (ранібізумаб у дозі 0,5 мг, режим дозування встановлювався за критеріями «активність захворювання», які визначалися як порушення зору, пов’язане з наявністю інтра- або субретинальної рідини або активним пропотіванням внаслідок ураження ХНВ, що оцінювалося за допомогою оптичної когерентної томографії та/або флуоресцентної ангіографії).
- Група III (вФДТ, пацієнтам було дозволено отримувати лікування ранібізумабом, починаючи з 3-го місяця).
У групі II, де застосовували рекомендовану дозу (див. розділ «Спосіб застосування та дози»), 50,9 % пацієнтів потребували 1 або 2 ін’єкцій, 34,5 % потребували від 3 до 5 ін’єкцій та 14,7 % потребували від 6 до 12 ін’єкцій протягом 12-місячного періоду дослідження. 62,9 % пацієнтів у групі ІІ не потребували ін’єкцій протягом наступного 6-місячного періоду дослідження.
Покращення зору супроводжувалося зниженням показників товщини в центральній ділянці сітківки (ТЦДС).
Пацієнти повідомляли про переваги лікування в групах застосування ранібізумабу порівняно із застосуванням вФДТ (р-значення < 0,05), які включали сумарну оцінку та показники декількох підшкал (загальний зір, зір на близькій відстані, психологічне здоров’я та залежність) в опитуванні NEI VFQ-25.
Лікування порушення зору внаслідок ХНВ (окрім тієї, що виникла внаслідок ПМ та ексудативної ВМД)
Клінічну безпеку та ефективність застосування ранібізумабу в пацієнтів із порушенням зору, спричиненим ХНВ, оцінювали на підставі даних, отриманих протягом 12 місяців у ході проведення рандомізованого, подвійно маскованого, плацебо-контрольованого базового дослідження G2301 (MINERVA). У цьому дослідженні 178 дорослих пацієнтів були рандомізовані у співвідношенні 2:1 для отримання:
- ранібізумабу в дозі 0,5 мг на початковому рівні з наступним індивідуальним режимом дозування, обумовленим активністю захворювання, що оцінюється за гостротою зору та/або анатомічними параметрами (наприклад, порушення ГЗ, наявність інтра/субретинальної рідини, крововилив або витік крові);
- ін’єкції плацебо на початковому рівні, за якою слідує індивідуальна схема лікування, обумовлена активністю захворювання.
Починаючи з 2-го місяця усі пацієнти отримували відкрите лікування ранібізумабом за потреби.
Покращення зору супроводжувалося зниженням показників ТЦДС протягом 12-місячного періоду.
Середня кількість ін’єкцій, отриманих протягом 12 місяців, становила 5,8 у групі лікування ранібізумабом порівняно з 5,4 у групі плацебо, пацієнти якої отримували ранібізумаб починаючи з 2-го місяця і надалі. У групі плацебо 7 із 59 пацієнтів не отримували ранібізумаб у хворе око протягом 12-місячного періоду.
Під час порівняння на 2-му місяці групи лікування ранібізумабом із контрольною групою плацебо спостерігався стійкий ефект лікування як у загальній групі, так і у підгрупах пацієнтів із захворюванням різних етіологій на початковому рівні.
У базовому дослідженні G2301 (MINERVA) п’ятеро підлітків віком від 12 до 17 років із порушенням зору, спричиненим ХНВ, отримували відкрите лікування ранібізумабом у дозі 0,5 мг на початковому рівні, а надалі – за індивідуальною схемою лікування як і для дорослих пацієнтів. Покращення показників МСГЗ від початкового рівня до 12-го місяця було досягнуто в усіх п’яти пацієнтів у межах від 5 до 38 літер (у середньому 16,6 літери). Покращення зору супроводжувалося стабілізацією або зниженням показників ТЦДС протягом 12-місячного періоду. Середня кількість ін’єкцій ранібізумабу, що були введені в досліджуване око протягом 12 місяців, становила 3 (від 2 до 5). Загалом, лікування ранібізумабом добре переносилося.
Лікування порушення зору при діабетичному макулярному набряку (ДМН)
Ефективність та безпеку застосування ранібізумабу оцінювали у ході трьох рандомізованих, контрольованих досліджень тривалістю щонайменше 12 місяців. Загалом у дослідження було залучено 868 пацієнтів (708 – в активну та 160 – у контрольну групи).
У дослідженні фази II D2201 (RESOLVE) 151 пацієнт отримував лікування ранібізумабом (6 мг/мл, n = 51, 10 мг/мл, n = 51) або плацебо (n = 49) у вигляді інтравітреальних ін’єкцій 1 раз на місяць. Середня зміна показника МСГЗ протягом періоду від 1-го до 12-го місяця порівняно з початковим рівнем становила + 7,8 (± 7,72) літери в об’єднаній групі пацієнтів, які отримували ранібізумаб (n = 102), порівняно з -0,1 (± 9,77) літери у пацієнтів, які отримували плацебо. Середня зміна показника МСГЗ на 12-му місяці від початкового рівня становила 10,3 (± 9,1) літери порівняно з -1,4 (± 14,2) літери відповідно (p < 0,0001 для відмінності між видами лікування).
У дослідженні фази III D2301 (RESTORE) 345 пацієнтів були рандомізовані у співвідношенні 1:1:1 для отримання ранібізумабу в дозі 0,5 мг як монотерапії та плацебо-лазерної фотокоагуляції, комбінованої терапії ранібізумабом у дозі 0,5 мг та лазерної фотокоагуляції або ін’єкції плацебо та лазерної фотокоагуляції. 240 пацієнтів, які раніше завершили 12-місячне дослідження RESTORE, були відібрані для участі у відкритому багатоцентровому 24-місячному розширеному дослідженні (розширене дослідження RESTORE). Пацієнти отримували лікування ранібізумабом у дозі 0,5 мг pro re nata (PRN – за потреби) у те ж саме досліджуване око, що й в основному дослідженні (D2301 RESTORE).
Ефект збігався у більшості підгруп на 12-му місяці. Однак у пацієнтів на початковому рівні з МСГЗ > 73 літери при макулярному набряку із товщиною в центральній ділянці сітківки < 300 мкм не спостерігалося користі від терапії ранібізумабом порівняно з лазерною фотокоагуляцією.
Статистично значуще покращення за повідомленнями пацієнтів стосовно більшості зорових функцій спостерігалося на фоні лікування ранібізумабом (з або без лазерної фотокоагуляції) порівняно з контрольною групою згідно з опитуванням NEI VFQ-25. За іншими показниками цього опитування відмінностей між видами лікування не було встановлено.
Профіль безпеки ранібізумабу при довготривалому застосуванні, що спостерігався у 24-місячному розширеному дослідженні, відповідає відомому профілю безпеки ранібізумабу.
У дослідженні фази IIIb D2304 (RETAIN) 372 пацієнти були рандомізовані у співвідношенні 1:1:1 для отримання:
- Ранібізумабу в дозі 0,5 мг із супутньою лазерною фотокоагуляцією за схемою «treat-and-extend» (ТЕ – «лікування та подовження»).
- Ранібізумабу в дозі 0,5 мг як монотерапії за схемою ТЕ.
- Ранібізумабу в дозі 0,5 мг як монотерапії за схемою PRN.
У всіх групах ранібізумаб вводили 1 раз на місяць і продовжували, доки МСГЗ не була стабільною протягом щонайменше трьох послідовних оцінювань 1 раз на місяць. За схемою ТЕ ранібізумаб вводили з інтервалами 2–3 місяці. У всіх групах терапія із застосуванням 1 раз на місяць розпочиналася повторно після зниження МСГЗ внаслідок прогресування ДМН та продовжувалася до повторного досягнення стабілізації показника МСГЗ.
Після перших трьох ін’єкцій кількість планованих лікувальних візитів становила 13 та 20 для схем ТЕ та PRN відповідно. В умовах застосування обох схем ТЕ у більш ніж 70 % пацієнтів зберігався рівень МСГЗ із середньою частотою візитів ≥ 2 місяці.
У дослідженнях ДМН покращення МСГЗ супроводжувалося зменшенням у динаміці середнього показника ТЦДС у всіх групах лікування.
Лікування проліферативної діабетичної ретинопатії (ПДР)
Клінічну безпеку та ефективність застосування ранібізумабу в пацієнтів із ПДР оцінювали в дослідженні Protocol S застосування інтравітреальних ін’єкцій ранібізумабу в дозі 0,5 мг порівняно з панретинальною фотокоагуляцією (ПФК). Первинною кінцевою точкою була середня зміна показника гостроти зору на 2-му році. Додатково динаміку тяжкості діабетичної ретинопатії (ДР) оцінювали за фотографіями очного дна з використанням шкали тяжкості діабетичної ретинопатії (DRSS).
Protocol S являв собою багатоцентрове, рандомізоване, із застосуванням активного контрольного лікарського засобу, паралельне дослідження для порівняння ефективності фази III, в якому брали участь 305 пацієнтів (394 хворих ока) із ПДР з або без ДМН на початковому рівні. У дослідженні порівнювали інтравітреальні ін’єкції ранібізумабу в дозі 0,5 мг зі стандартним лікуванням ПФК. Загалом 191 око (48,5 %) було рандомізовано для введення ранібізумабу в дозі 0,5 мг, а 203 ока (51,5 %) було рандомізовано для ПФК. Загалом у 88 очах (22,3 %) було діагностовано ДМН на початковому рівні: 42 (22,0 %) і 46 (22,7 %) очей у групах лікування ранібізумабом і ПФК відповідно.
У цьому дослідженні середня зміна показника гостроти зору протягом 2-х років становила +2,7 літери у групі лікування ранібізумабом порівняно з -0,7 літери у групі лікування ПФК. Різниця в середніх значеннях найменших квадратів становила 3,5 літери (95 % ДІ: [0,2–6,7]).
Протягом 1-го року в 41,8 % очей було відзначено покращення на ≥ 2 ступеня за шкалою DRSS при лікуванні ранібізумабом (n = 189) порівняно з 14,6 % очей, лікування яких здійснювали за допомогою ПФК (n = 199). Оцінка результатів розбіжностей між ранібізумабом та лазерною фотокоагуляцією становила 27,4 % (95 % ДІ: [18,9; 35,9]).
Протягом 1-го року в групі лікування ранібізумабом дослідження Protocol S ≥ 2-ступеневе покращення за шкалою DRSS було стійким в очах без ДМН (39,9 %) та з ДМН на початковому рівні (48,8 %).
За результатами аналізу даних, одержаних протягом 2 років у дослідженні Protocol S, було визначено покращення на ≥ 2 ступені за шкалою DRSS у 42,3 % (n = 80) очей у групі лікування ранібізумабом від початкового рівня порівняно з 23,1 % (n = 46) очей у групі лікування ПФК. У групі лікування ранібізумабом було визначено покращення на ≥ 2 ступені за шкалою DRSS від початкового рівня у 58,5 % (n = 24) очей з ДМН початкового рівня та 37,8 % (n = 56) очей без ДМН.
DRSS також оцінювали у трьох окремих дослідженнях ДМН фази III з активним контрольним лікарським засобом (ранібізумаб у дозі 0,5 мг за схемою PRN порівняно з ПФК), що включали загалом 875 пацієнтів, серед яких приблизно 75 % були азіатського походження. У метааналізі цих досліджень у 48,4 % із 315 пацієнтів з оцінками за шкалою DRSS у підгрупі пацієнтів із помірно тяжкою непроліферативною діабетичною ретинопатією (нПДР) або більш тяжкою на початковому рівні спостерігалося покращення на ≥ 2 ступені за шкалою DRSS протягом 12 місяців при лікуванні ранібізумабом (n = 192) порівняно з 14,6 % пацієнтів, які отримували лікування лазерною фотокоагуляцією (n = 123). Оцінка результатів розбіжностей між ранібізумабом та лазерною фотокоагуляцією становила 29,9 % (95 % ДІ: [20,0; 39,7]). У 405 пацієнтів з оцінкою за шкалою DRSS із помірною або легкою нПДР покращення на ≥ 2 ступені за шкалою DRSS спостерігалося у 1,4 % та 0,9 % в групах лікування ранібізумабом і лазерною фотокоагуляцією відповідно.
Лікування порушення зору при макулярному набряку, що виник внаслідок тромбозу вен сітківки (ТВС)
Клінічну безпеку та ефективність застосування ранібізумабу в пацієнтів із порушенням зору при макулярному набряку, що виник внаслідок ТВС, оцінювали в рандомізованих, подвійно маскованих, контрольованих дослідженнях BRAVO та CRUISE за участю пацієнтів із тромбозом гілок центральної вени сітківки (ТГЦВС) (n = 397) та тромбозом центральної вени сітківки (ТЦВС) (n = 392) відповідно. В обох дослідженнях пацієнтам вводили ранібізумаб у дозі 0,3 мг або 0,5 мг, або плацебо. Через 6 місяців пацієнтів контрольної групи плацебо переводили на лікування ранібізумабом у дозі 0,5 мг.
В обох дослідженнях покращення зору супроводжувалося постійним та значним зменшенням макулярного набряку, виміряного за ТЦДС.
У пацієнтів із ТЦВС (CRUISE та розширене дослідження HORIZON): пацієнти контрольної групи, які отримували плацебо протягом перших 6 місяців, а в подальшому були переведені на лікування ранібізумабом, покращення ГЗ до 24-го місяця (~6 літер) не досягло рівня, подібного до відзначеного у тих, хто отримував ранібізумаб від початку дослідження (~12 літер).
Статистично значуще покращення за повідомленнями пацієнтів за показниками підшкал оцінки ближнього та дальнього зору спостерігалося при лікуванні ранібізумабом порівняно з контрольною групою в опитуванні NEI VFQ-25.
Довготривала (24 місяці) клінічна безпека та ефективність ранібізумабу в пацієнтів із порушенням зору при макулярному набряку, що виник внаслідок ТВС, оцінювались у ході досліджень BRIGHTER (ТГЦВС) та CRYSTAL (ТЦВС). В обох дослідженнях пацієнтам призначали лікування за схемою дозування PRN ранібізумабу в дозі 0,5 мг, що встановлювалася за індивідуальними критеріями стабільності. BRIGHTER – рандомізоване, активно контрольоване дослідження в трьох групах для порівняння застосування ранібізумабу в дозі 0,5 мг як монотерапії або в комбінації з ад’ювантною лазерною фотокоагуляцією порівняно з лазерною фотокоагуляцією окремо. Через 6 місяців пацієнти групи лазерної фотокоагуляції могли отримувати ранібізумаб у дозі 0,5 мг. CRYSTAL – непорівняльне дослідження застосування ранібізумабу в дозі 0,5 мг як монотерапії.
У дослідженні BRIGHTER ранібізумаб у дозі 0,5 мг з ад’ювантною лазерною терапією продемонстрував не вищу ефективність порівняно із застосуванням ранібізумабу як монотерапії від початкового рівня до 24-го місяця (95 % ДІ: -2,8; 1,4).
В обох дослідженнях швидке та статистично значуще зменшення показників ТЦДС від початкового рівня спостерігалося на 1-му місяці. Цей ефект зберігався до 24-го місяця.
Ефект лікування ранібізумабом був аналогічним, незважаючи на наявність ішемії сітківки. У дослідженні BRIGHTER у пацієнтів з ішемією (n = 46) або без неї (n = 133), які застосовували ранібізумаб як монотерапію, середня зміна від початкового рівня становила +15,3 та +15,6 літери відповідно на 24-му місяці. У дослідженні CRYSTAL у пацієнтів з ішемією (n = 53) або без неї (n = 300), які застосовували ранібізумаб як монотерапію, середня зміна від початкового рівня становила +15,0 та +11,5 літер відповідно.
Під час досліджень BRIGHTER та CRYSTAL покращення зору з часом спостерігалося в усіх пацієнтів, які застосовували ранібізумаб у дозі 0,5 мг як монотерапію, незважаючи на тривалість захворювання. У пацієнтів із тривалістю захворювання < 3 місяців спостерігалося покращення гостроти зору на 13,3 та 10,0 літери на 1-му місяці та на 17,7 і 13,2 літери на 24-му місяці в ході досліджень BRIGHTER та CRYSTAL відповідно. Відповідне покращення гостроти зору в пацієнтів з тривалістю захворювання > 12 місяців становило 8,6 та 8,4 літери у відповідних дослідженнях. Слід розглянути можливість початку лікування при діагностуванні захворювання.
Профіль довготривалої безпеки ранібізумабу, що спостерігався у 24-місячних дослідженнях, відповідає відомому профілю безпеки ранібізумабу.
Діти
Європейське агентство з лікарських засобів відклало вимогу щодо надання результатів досліджень із ранібізумабом у всіх підгрупах пацієнтів дитячого віку при неоваскулярній ВМД, порушенні зору при ДМН, порушенні зору при макулярному набряку, що виник внаслідок ТВС, та порушенні зору внаслідок ХНВ та діабетичної ретинопатії (див. розділ «Спосіб застосування та дози» для отримання інформації про застосування дітям).
Фармакокінетика.
Після інтравітреального введення ранібізумабу (1 раз на місяць) у пацієнтів із неоваскулярною ВМД концентрація ранібізумабу в сироватці крові була, як правило, низькою, з максимальним рівнем (Cmax), зазвичай нижчим за концентрацію ранібізумабу, необхідну для інгібування біологічної активності судинного ендотеліального фактора росту (VEGF) на 50 % (11–27 нг/мл за даними аналізу клітинної проліферації in vitro). Cmax була пропорційна дозі в діапазоні доз 0,05–1,0 мг/око. Сироваткові концентрації у деяких пацієнтів з ДМН показують, що не можна виключити дещо вищу системну експозицію порівняно з тією, що спостерігається в пацієнтів з неоваскулярною ВМД. Сироваткові концентрації ранібізумабу в пацієнтів з ТВС були подібними до тих, що спостерігалися в пацієнтів із неоваскулярною ВМД, або дещо їх перевищували.
Згідно з даними популяційної фармакокінетики і виведення ранібізумабу із сироватки крові у пацієнтів із неоваскулярною ВМД, які отримували ранібізумаб у дозі 0,5 мг, середній період напіввиведення ранібізумабу зі склоподібного тіла становить приблизно 9 днів. При інтравітреальному введенні ранібізумабу в дозі 0,5 мг/око (1 раз на місяць) Cmax ранібізумабу в сироватці крові спостерігається приблизно через 1 день після введення і зазвичай становить від 0,79–2,90 нг/мл, Cmin може становити від 0,07–0,49 нг/мл. Експозиція ранібізумабу в сироватці крові приблизно в 90 000 разів нижча, ніж у склоподібному тілі.
Пацієнти з порушенням функції нирок
Офіційні дослідження фармакокінетики ранібізумабу за участю пацієнтів із порушенням функції нирок не проводили. При вивченні фармакокінетики у групі пацієнтів із неоваскулярною ВМД 68 % (136 із 200) пацієнтів мали порушення функції нирок (46,5 % незначні [50–80 мл/хв], 20 % помірні [30–50 мл/хв] та 1,5 % тяжкі [< 30 мл/хв]). У групі пацієнтів із ТВС 48,2 % (253 із 525) пацієнтів мали порушення функції нирок (36,4 % незначні, 9,5 % помірні та 2,3 % тяжкі). Рівень системного кліренсу був дещо нижчий, але клінічно незначущий.
Пацієнти із порушенням функції печінки
Офіційні дослідження фармакокінетики ранібізумабу за участю пацієнтів із порушенням функції печінки не проводили.
Клінічні характеристики.
Показання.
- Неоваскулярна (ексудативна) вікова макулярна дегенерація (ВМД).
- Порушення зору при діабетичному макулярному набряку (ДМН).
- Проліферативна діабетична ретинопатія (ПДР).
- Порушення зору при макулярному набряку, що виник внаслідок тромбозу вен сітківки (тромбоз гілок центральної вени сітківки (ТГЦВС) або тромбоз центральної вени сітківки (ТЦВС)).
- Порушення зору внаслідок хоріоїдальної неоваскуляризації (ХНВ).
Протипоказання.
− Підвищена чутливість до діючої речовини або до будь-якої з допоміжних речовин препарату.
− Активна або підозрювана окулярна, або періокулярна інфекція.
− Активне тяжке внутрішньоочне запалення.
Взаємодія з іншими лікарськими засобами та інші види взаємодій.
Офіційні дослідження взаємодії не проводили.
Інформацію щодо одночасного застосування фотодинамічної терапії вертепорфіном (вФДТ) та ранібізумабу при ексудативній ВМД та ПМ див. у підрозділі «Фармакодинаміка».
Інформацію щодо одночасного застосування лазерної фотокоагуляції та ранібізумабу при ДМН та ТГЦВС див. у розділі «Спосіб застосування та дози» та у підрозділі «Фармакодинаміка».
У клінічних дослідженнях лікування порушення зору при ДМН результат стосовно гостроти зору або товщини в центральній ділянці сітківки (ТЦДС) у пацієнтів, які отримували лікування ранібізумабом, не залежав від одночасного лікування тіазолідиндіоном.
Особливості застосування.
Відстежуваність
З метою покращення відстежуваності біологічних лікарських засобів потрібно чітко записувати назву та номер серії лікарського засобу, що застосовується.
Реакції, пов’язані з інтравітреальною ін’єкцією
Інтравітреальні ін’єкції, включаючи ін’єкції ранібізумабу, були пов’язані з ендофтальмітом, внутрішньоочним запаленням, регматогенним відшаруванням сітківки ока, розривом сітківки та ятрогенною травматичною катарактою (див. розділ «Побічні реакції»). Ін’єкцію ранібізумабу слід завжди проводити в належних асептичних умовах. Крім того, потрібно спостерігати за станом пацієнта протягом тижня після ін’єкції з метою своєчасного лікування у разі розвитку інфекційного ускладнення. Слід інформувати пацієнтів про необхідність негайно повідомляти лікаря про появу будь-яких симптомів, які можуть нагадувати ендофтальміт або інші вищезазначені стани.
Підвищення внутрішньоочного тиску (ВОТ)
Транзиторне підвищення ВОТ відзначали протягом 60 хвилин після ін’єкції ранібізумабу. Також виявляли стійке підвищення ВОТ (див. розділ «Побічні реакції»). Показники ВОТ та перфузії диска зорового нерва слід перевірити і відповідно скоригувати.
Пацієнтів слід проінформувати про симптоми цих потенційних побічних реакцій та про необхідність повідомляти свого лікаря про появу будь-яких ознак, зокрема таких, як біль у очах або підвищений дискомфорт, посилення почервоніння очей, помутніння або зниження гостроти зору, збільшення кількості дрібних частинок у полі зору або підвищена чутливість до світла (див. розділ «Побічні реакції»).
Білатеральна терапія
Обмежені дані щодо білатерального застосування ранібізумабу (включаючи введення в один і той же день) свідчать про відсутність підвищеного ризику системних небажаних побічних явищ порівняно з одностороннім введенням.
Імуногенність
Існує ризик виникнення імуногенності при застосуванні ранібізумабу. Оскільки у пацієнтів із ДМН існує ймовірність збільшення системної експозиції, такі пацієнти мають підвищений ризик розвитку реакції гіперчутливості.
Пацієнтів також слід попередити про необхідність повідомляти про всі симптоми, які свідчать про розвиток внутрішньоочного запалення, що може бути клінічною ознакою утворення внутрішньоочних антитіл.
Одночасне застосування з іншими інгібіторами VEGF (судинний ендотеліальний фактор росту)
Не слід одночасно застосовувати ранібізумаб з іншими інгібіторами VEGF (при введенні препарату в системний кровообіг або око).
Припинення застосування ранібізумабу
Введення ранібізумабу можна припинити та не поновлювати раніше наступного запланованого введення у разі:
- зниження максимально скоригованої гостроти зору (МСГЗ) на ≥ 30 літер порівняно з останніми показниками перевірки гостроти зору;
- внутрішньоочного тиску ≥ 30 мм рт. ст.;
- розриву сітківки;
- субретинального крововиливу, який досягнув центру ямки сітківки або якщо розмір крововиливу становить ≥ 50 % загальної площі ураження;
- виконаного або запланованого хірургічного втручання на очах протягом 28 днів до або після ін’єкції.
Розрив пігментного епітелію сітківки
Фактори ризику, пов’язані з розвитком розриву пігментного епітелію сітківки після застосування інгібіторів VEGF для лікування ексудативної ВМД та потенційно іншими формами ХНВ, включають широке та/або високе відшарування пігментного епітелію сітківки. На початку терапії ранібізумабом слід бути обережними пацієнтам із факторами ризику розривів пігментного епітелію сітківки.
Регматогенне відшарування сітківки або макулярні отвори
Лікування слід припинити пацієнтам із регматогенним відшаруванням сітківки або із макулярними отворами 3–4-го ступеня.
Групи пацієнтів, досвід застосування препарату яким обмежений
Існує обмежений досвід лікування пацієнтів із ДМН внаслідок цукрового діабету I типу. Не досліджували застосування ранібізумабу пацієнтам, яким раніше вводили інтравітреальні ін’єкції, пацієнтам із активними системними інфекціями або пацієнтам із супутніми офтальмологічними захворюваннями, такими як відшарування сітківки або макулярні отвори. Також відсутній досвід лікування ранібізумабом пацієнтів із цукровим діабетом, у яких рівень глікозильованого гемоглобіну HbA1c понад 108 ммоль/моль (12 %) та пацієнтів із неконтрольованою гіпертензією. При лікуванні таких пацієнтів лікар повинен враховувати вищезазначену інформацію.
Існує недостатньо даних, щоб зробити висновок щодо ефективності застосування ранібізумабу пацієнтам із ТВС, в яких спостерігається необоротна втрата зорових функцій внаслідок ішемії.
Існують обмежені дані щодо дії ранібізумабу в пацієнтів із ПМ, які не відповіли на попередню фотодинамічну терапію вертепорфіном (вФДТ). Хоча у пацієнтів із субфовеальними та юкстафовеальними ураженнями спостерігався стійкий ефект, даних для висновку щодо дії ранібізумабу в пацієнтів із ПМ та екстрафовеальними ураженнями недостатньо.
Системні ефекти після інтравітреального застосування
Системні побічні явища, в тому числі позаочні крововиливи та артеріальні тромбоемболічні явища, іноді спостерігалися після інтравітреальних ін’єкцій інгібіторів VEGF.
Існують обмежені дані щодо безпеки лікування пацієнтів із ДМН, макулярним набряком внаслідок ТВС та ХНВ, що виникла внаслідок ПМ, а також пацієнтів, які мають в анамнезі інсульт або транзиторні ішемічні атаки. Слід бути обережними під час лікування таких пацієнтів (див. розділ «Побічні реакції»).
Застосування у період вагітності або годування груддю.
Жінки репродуктивного віку/контрацепція у жінок
Жінки репродуктивного віку повинні застосовувати ефективні методи контрацепції під час лікування.
Вагітність
Клінічні дані щодо впливу ранібізумабу вагітним відсутні. Дослідження на мавпах роду Сynomolgus не свідчать про прямий або опосередкований несприятливий вплив на перебіг вагітності або розвиток ембріона/плода. Системна експозиція ранібізумабу після внутрішньоочного введення низька, але з огляду на механізм дії ранібізумаб необхідно розглядати як потенційно тератогенний та ембріо-/фетотоксичний препарат. Тому ранібізумаб не рекомендується застосовувати у період вагітності, за винятком випадків, коли очікувана користь для жінки перевищує потенційний ризик для плода. Жінкам, які планують вагітність та яким вводили ранібізумаб, рекомендовано, щоб між останнім введенням препарату та зачаттям дитини минуло щонайменше 3 місяці.
Годування груддю
Невідомо, чи екскретується ранібізумаб у грудне молоко. Тому годування груддю не рекомендоване під час застосування препарату.
Фертильність
Дані щодо впливу на фертильність відсутні.
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами.
Лікування препаратом може спричинити тимчасове порушення зору, що, у свою чергу, може впливати на здатність керувати автотранспортом або іншими механізмами (див. розділ «Побічні реакції»). Пацієнтам, які відзначали такі симптоми, не слід керувати автотранспортом та іншими механізмами до зникнення вищезазначених тимчасових симптомів.
Спосіб застосування та дози.
Тільки для інтравітреального введення. Введення лікарського засобу АМЕЛІВУ повинен виконувати тільки кваліфікований лікар-офтальмолог, який має досвід проведення інтравітреальних ін’єкцій.
Дозування
Рекомендована доза лікарського засобу АМЕЛІВУ становить 0,5 мг у вигляді одноразової інтравітреальної ін’єкції. Ця доза відповідає об’єму ін’єкції 0,05 мл. Інтервал між введеннями двох доз в одне око повинен становити щонайменше 4 тижні.
Лікування слід розпочинати з 1 ін’єкції на місяць і продовжувати до досягнення максимальної гостроти зору та/або зникнення ознак активності захворювання, тобто відсутності зміни гостроти зору та інших ознак і симптомів захворювання на фоні продовження лікування. Пацієнтам із ексудативною ВМД, ДМН, ПДР та ТВС спочатку може бути потрібно 3 або більше послідовних ін’єкцій з частотою 1 раз на місяць.
Таким чином, регулярність моніторингу та інтервали між введеннями препарату має визначати лікар залежно від активності захворювання, що оцінюється на основі гостроти зору та/або за анатомічними параметрами.
Якщо, на думку лікаря, показники гостроти зору та анатомічні параметри вказують на відсутність користі від продовження лікування для пацієнта, застосування лікарського засобу АМЕЛІВУ слід припинити.
Моніторинг активності захворювання може включати клінічне обстеження, функціональні проби або методи візуалізації (наприклад, оптична когерентна томографія або флуоресцентна ангіографія).
Якщо пацієнти отримують лікування за схемою «treat-and-extend» (лікування та подовження) після досягнення максимальної гостроти зору та/або за відсутності ознак активності захворювання, інтервали між введеннями препарату можна поступово подовжити до рецидиву ознак активності захворювання або порушення зору. Інтервали між введеннями препарату можна подовжувати не більше ніж на 2 тижні одноразово у випадку ексудативної ВМД і не більше ніж на 1 місяць одноразово у випадку ДМН. У випадку ПДР та ТВС інтервали між введеннями препарату також можна поступово подовжувати, однак доступних даних недостатньо для визначення тривалості цих інтервалів. У разі рецидиву активності захворювання інтервали між введеннями препарату слід відповідно скоротити.
Лікування порушення зору внаслідок ХНВ слід призначати індивідуально для кожного пацієнта на основі активності захворювання. Деяким пацієнтам може бути достатньо тільки однієї ін’єкції протягом перших 12 місяців; інші пацієнти можуть потребувати більш частого введення препарату, включаючи ін’єкцію 1 раз на місяць. Якщо лікування проводиться з приводу ХНВ, що виникла внаслідок патологічної міопії (ПМ), більшість пацієнтів може потребувати тільки однієї або двох ін’єкцій протягом першого року (див. підрозділ «Фармакодинаміка»).
Застосування ранібізумабу та лазерної фотокоагуляції при ДМН та макулярному набряку, що виник внаслідок тромбозу гілок центральної вени сітківки (ТГЦВС)
Існує невеликий досвід введення ранібізумабу одночасно з лазерною фотокоагуляцією (див. підрозділ «Фармакодинаміка»). При застосуванні в один і той самий день ранібізумаб потрібно вводити щонайменше через 30 хвилин після лазерної фотокоагуляції. Ранібізумаб можна застосовувати пацієнтам, яким раніше проводили лазерну фотокоагуляцію.
Застосування ранібізумабу та вФДТ при ХНВ, що виникла внаслідок ПМ
Дані щодо одночасного застосування ранібізумабу та вертепорфіну відсутні.
Особливі групи пацієнтів
Пацієнти із порушенням функції печінки
Не досліджували застосування ранібізумабу пацієнтам із порушенням функції печінки. Проте ця група пацієнтів не потребує особливої уваги.
Пацієнти із порушенням функції нирок
Пацієнтам із порушенням функції нирок коригування дози не потрібне (див. підрозділ «Фармакокінетика»).
Пацієнти літнього віку
Пацієнтам літнього віку коригування дози не потрібне. Досвід застосування пацієнтам віком від 75 років із ДМН обмежений.
Спосіб введення
Оскільки об’єм, що міститься у флаконі (0,23 мл), перевищує рекомендовану дозу (0,05 мл), частину об’єму, що міститься у флаконі, слід утилізувати перед введенням.
Перед введенням препарат АМЕЛІВУ потрібно візуально перевірити на наявність твердих частинок та зміну кольору.
Процедуру введення препарату проводити в асептичних умовах, які включають: хірургічну дезінфекцію рук, використання стерильних рукавичок, стерильного простирадла, стерильного розширювача повік (або еквівалентний пристрій) та стерильного інструменту для парацентезу (за необхідності). Слід уважно переглянути медичний анамнез пацієнта на наявність реакцій гіперчутливості перед проведенням інтравітреальної ін’єкції (див. розділ «Особливості застосування»). Перед проведенням ін’єкції потрібно визначити відповідну анестезію та бактерицидний засіб широкого спектра дії для дезінфекції шкіри навколо ока, повік та поверхні ока згідно з місцевою практикою.
Голку для ін’єкцій потрібно ввести на 3,5–4,0 мм позаду від лімба у склоподібне тіло, відхиляючись від горизонтального меридіана та спрямовуючи голку у напрямі до центра очного яблука. Потім ввести 0,05 мл розчину; місце проколу склери потрібно змінювати при наступних ін’єкціях.
Флакон призначений тільки для одноразового використання. Після ін’єкції будь-яку кількість невикористаного препарату слід утилізувати. Не використовувати флакон, якщо його упаковка має ознаки пошкодження або порушення цілісності. У випадку порушення герметичності упаковки стерильність не гарантується.
Для приготування та інтравітреального введення слід використовувати такі медичні вироби для одноразового використання:
- голка з фільтром на 5 мкм (18G);
- голка для ін’єкцій (30G × ½");
- стерильний шприц об’ємом 1 мл (який має поділку 0,05 мл).
Ці медичні вироби не входять до комплекту цієї упаковки.
Більш детальна інструкція щодо підготовки лікарського засобу АМЕЛІВУ для інтравітреального введення наведена нижче.
Для підготовки лікарського засобу АМЕЛІВУ для інтравітреального введення слід дотримуватися нижчезазначених інструкцій.
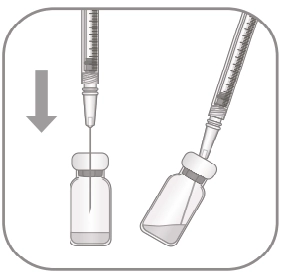
|
1. Перед забором вмісту флакона зовнішню частину гумової пробки необхідно продезінфікувати. 2. З’єднати голку з фільтром на 5 мкм (18G × 1½", 1,2 мм × 40 мм) зі шприцом об’ємом 1 мл в асептичних умовах. Ввести голку з фільтром у центральну частину гумової пробки флакона, поки голка не торкнеться дна флакона. 3. Забрати всю рідину з флакона, тримаючи флакон вертикально і трохи нахиливши в бік для більш зручного повного забору вмісту. |
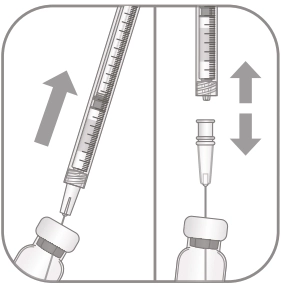
|
4. Переконатись, що шток поршня повністю відведений назад при спорожненні флакона, щоб повністю випорожнити голку з фільтром. 5. Залишити голку з фільтром у флаконі та від’єднати шприц від голки з фільтром. Голку з фільтром слід утилізувати після забору вмісту флакона, її не слід використовувати для інтравітреального введення. |
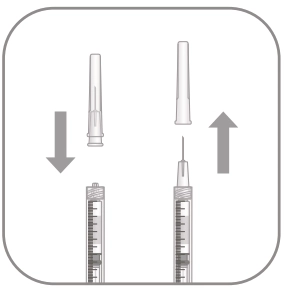
|
6. В асептичних умовах щільно приєднати голку для ін’єкцій (30G × ½", 0,3 мм × 13 мм) до шприца. 7. Обережно зняти ковпачок з голки для ін’єкцій так, щоб не роз’єднати голку зі шприцом. Примітка: знімаючи ковпачок голки для ін’єкцій, тримайте основу голки. |
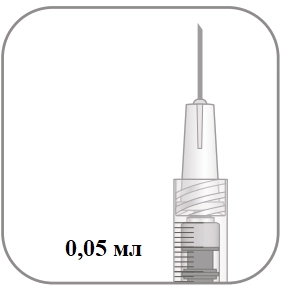
|
8. Обережно витиснути повітря зі шприца разом із надлишком розчину та відрегулювати дозу на рівні поділки 0,05 мл на шприці. Шприц готовий для проведення ін’єкції. Примітка: не протирати голку для ін’єкцій. Не відводити поршень шприца назад. |
Після ін’єкції не надягати знову ковпачок на голку та не від’єднувати її від шприца. Використаний шприц разом із голкою слід утилізувати в контейнер для використаних гострих предметів або відповідно до вимог чинного законодавства.
Діти.
Безпека та ефективність застосування ранібізумабу дітям та підліткам (віком до 18 років) не встановлені. Доступні дані щодо застосування ранібізумабу підліткам віком від 12 до 17 років із порушенням зору внаслідок ХНВ описано в підрозділі «Фармакодинаміка», однак рекомендації щодо дозування відсутні.
Передозування.
Про випадкове передозування повідомлялося під час клінічних досліджень ексудативної ВМД та у період післяреєстраційного застосування. Побічні реакції, найчастіше пов’язані з передозуванням: підвищення внутрішньоочного тиску, транзиторна сліпота, зниження гостроти зору, набряк рогівки, біль у рогівці та біль в оці. При передозуванні необхідно стежити за рівнем внутрішньоочного тиску і у разі потреби розпочати відповідне лікування, якщо лікар вважає за необхідне.
Побічні реакції.
Більшість побічних реакцій, про які повідомлялося під час застосування ранібізумабу, пов’язані з процедурою інтравітреальної ін’єкції.
Найчастіші побічні реакції, пов’язані з процедурою введення ранібізумабу, включали: біль в оці, гіперемію ока, підвищення внутрішньоочного тиску, запалення склоподібного тіла, відшарування склоподібного тіла, крововиливи в сітківку, порушення зору, плаваючі помутніння склоподібного тіла, крововиливи в кон’юнктиву, подразнення ока, відчуття стороннього тіла в оці, підвищене сльозовиділення, блефарит, сухість очей та відчуття свербежу в оці.
Найчастіше повідомлялося про такі неофтальмологічні побічні реакції, як головний біль, назофарингіт та артралгія.
Менш часті, але більш тяжкі побічні реакції включали ендофтальміт, сліпоту, відшарування сітківки, розрив сітківки та ятрогенну травматичну катаракту (див. розділ «Особливості застосування»).
Побічні реакції, що спостерігалися після введення ранібізумабу у клінічних дослідженнях, представлені в таблиці нижче.
Побічні реакції# наведено за класами систем органів та частотою виникнення згідно з такою систематизацією: дуже часті (≥ 1/10), часті (від ≥ 1/100 до < 1/10), нечасті (від ≥ 1/1000 до < 1/100), поодинокі (від ≥ 1/10 000 до < 1/1000), рідкісні (< 1/10 000), частота невідома (частоту не можна визначити з доступних даних). У кожній групі побічні реакції представлені у порядку зменшення їхньої серйозності.
|
Інфекції та інвазії |
|
|
Дуже часті |
Назофарингіт |
|
Часті |
Інфекція сечовивідних шляхів* |
|
З боку крові та лімфатичної системи |
|
|
Часті |
Анемія |
|
З боку імунної системи |
|
|
Часті |
Гіперчутливість |
|
Психічні розлади |
|
|
Часті |
Тривожність |
|
З боку нервової системи |
|
|
Дуже часті |
Головний біль |
|
З боку органів зору |
|
|
Дуже часті |
Запалення склоподібного тіла, відшарування склоподібного тіла, крововиливи в сітківку, порушення зору, біль в оці, плаваючі помутніння склоподібного тіла, крововиливи в кон’юнктиву, подразнення ока, відчуття стороннього тіла в оці, підвищене сльозовиділення, блефарит, сухість очей, гіперемія ока, відчуття свербежу в оці |
|
Часті |
Дегенерація сітківки, ураження сітківки, відшарування сітківки, розрив сітківки, відшарування пігментного епітелію сітківки, розрив пігментного епітелію сітківки, зниження гостроти зору, крововиливи в склоподібне тіло, ураження склоподібного тіла, увеїт, ірит, іридоцикліт, катаракта, субкапсулярна катаракта, помутніння задньої капсули, точковий кератит, ерозія рогівки, запалення передньої камери ока, порушення чіткості зору, крововиливи у місці ін’єкції, крововиливи в око, кон’юнктивіт, алергічний кон’юнктивіт, виділення з ока, фотопсія, фотофобія, відчуття дискомфорту в оці, набряк повіки, біль у повіці, гіперемія кон’юнктиви |
|
Нечасті |
Сліпота, ендофтальміт, гіпопіон, гіфема, кератопатія, спайки райдужки, відкладення на рогівці, набряк рогівки, утворення стрій на рогівці, біль у місці введення, подразнення у місці введення, незвичні відчуття в оці, подразнення повіки |
|
З боку дихальної системи, органів грудної клітки та середостіння |
|
|
Часті |
Кашель |
|
З боку шлунково-кишкового тракту |
|
|
Часті |
Нудота |
|
З боку шкіри та підшкірної клітковини |
|
|
Часті |
Алергічні реакції (висипання, кропив’янка, свербіж, еритема) |
|
З боку кістково-м’язової системи та сполучної тканини |
|
|
Дуже часті |
Артралгія |
|
Лабораторні показники |
|
|
Дуже часті |
Підвищення внутрішньоочного тиску |
|
# Побічні реакції визначали як побічні явища (щонайменше у 0,5 % пацієнтів), що виникали з більшою частотою (щонайменше у 2 %) у пацієнтів, які отримували лікування ранібізумабом у дозі 0,5 мг, порівняно з тими, хто отримував контрольний препарат (плацебо або вФДТ). * Спостерігалося тільки у пацієнтів із ДМН. |
|
Побічні реакції, пов’язані з класом препарату
У дослідженнях фази ІІІ ексудативної ВМД загальна частота неокулярних крововиливів, побічного явища, потенційно пов’язаного із застосуванням системних інгібіторів VEGF (судинного ендотеліального фактора росту), була дещо вищою у пацієнтів, які отримували ранібізумаб. Проте закономірності серед різних видів крововиливів не було. Існує теоретичний ризик появи артеріальних тромбоемболічних ускладнень після інтравітреального введення інгібіторів VEGF. У клінічних дослідженнях ранібізумабу в пацієнтів із ВМД, ДМН, ПДР, ТВС та ХНВ спостерігалася низька частота появи артеріальних тромбоемболічних ускладнень і не було суттєвих відмінностей між групами, які отримували ранібізумаб та контрольний препарат.
Повідомлення про підозрювані побічні реакції
Повідомлення про підозрювані побічні реакції після реєстрації лікарського засобу має важливе значення. Це дає змогу проводити безперервний моніторинг співвідношення між користю й ризиками, пов’язаними із застосуванням цього лікарського засобу. Медичним та фармацевтичним працівникам, а також пацієнтам або їх законним представникам слід повідомляти про усі випадки підозрюваних побічних реакцій та відсутності ефективності лікарського засобу через Автоматизовану інформаційну систему з фармаконагляду за посиланням: https://aisf.dec.gov.ua/.
Термін придатності.
30 місяців.
Умови зберігання.
Зберігати в холодильнику (при температурі від 2 до 8 °С) в оригінальній упаковці для захисту від дії світла. Не заморожувати.
Зберігати в недоступному для дітей місці та поза полем зору дітей.
Перед застосуванням невідкритий флакон можна зберігати при температурі не вище 30 °С протягом 1 місяця.
Несумісність.
Оскільки дослідження несумісності лікарського засобу не проводили, цей лікарський засіб не можна змішувати з іншими лікарськими засобами.
Упаковка.
Флакон з боросилікатного скла типу І з гумовою пробкою та алюмінієвим ковпачком з кришкою flip-off; по 1 флакону в стандартно-експортній упаковці в картонній коробці.
Категорія відпуску.
За рецептом.
Виробник.
Самсунг Біоепіс НЛ Б.В.
Місцезнаходження виробника та адреса місця провадження його діяльності.
Олоф Палмештраат 10, Делфт, 2616 LR, Нідерланди.
Заявник.
САМСУНГ БІОЕПІС КО., ЛТД.
Місцезнаходження заявника.
76, Сондогйоюк-ро, Йонсугу, Інчхон, Республіка Корея.