
АПРЕТЮД
-
Велика Британія ВііВ Хелскер ЮК ЛімітедФорма выпуску: суспензія для ін'єкцій пролонгованої дії, 200 мг/мл; по 3 мл у флаконі; по 1 або 25 флаконів в картонній коробціСклад: 1 флакон містить 600 мг каботегравіруУмови відпуску: за рецептомНомер реєстраційного посвідчення: UA/20557/01/01код АТС: J05AJ04Термін придатності: 3 роки
-
Велика Британія ВііВ Хелскер ЮК ЛімітедФорма выпуску: таблетки, вкриті плівковою оболонкою, по 30 мг; по 30 таблеток у флаконі; по 1 флакону в картонній коробціСклад: 1 таблетка містить 30 мг каботегравіру (у формі каботегравіру натрію)Умови відпуску: за рецептомНомер реєстраційного посвідчення: UA/20557/02/01код АТС: J05AJ04Термін придатності: 5 років
ІНСТРУКЦІЯ
для медичного застосування лікарського засобу
АПРЕТЮД
(APRETUDE)
Склад:
діюча речовина: каботегравір;
1 флакон містить 600 мг каботегравіру;
допоміжні речовини: маніт (Е 421), полісорбат 20, макрогол 3350, вода для ін’єкцій.
Лікарська форма. Суспензія для ін’єкцій пролонгованої дії.
Основні фізико-хімічні властивості: легкоплинна суспензія від білого до світло-рожевого кольору.
Фармакотерапевтична група. Противірусні лікарські засоби. Інгібітори інтегрази. Каботегравір. Код АТХ J05A J04.
Фармакологічні властивості.
Фармакодинаміка.
Механізм дії
Каботегравір інгібує інтегразу ВІЛ шляхом зв’язування з активним центром інтегрази та блокування ланцюга інтеграції ретровірусної дезоксирибонуклеїнової кислоти (ДНК) на етапі перенесення, що має вирішальне значення для циклу реплікації ВІЛ.
Фармакодинамічні ефекти
Противірусна активність у культурі клітин
Каботегравір виявляв противірусну активність щодо лабораторних штамів ВІЛ-1 дикого типу, при цьому середня концентрація каботегравіру, необхідна для зниження реплікації вірусу на 50 відсотків (EC50), становила 0,22 нМ у мононуклеарних клітинах периферичної крові (МКПК), 0,74 нМ у клітинах 293T та 0,57 нМ у клітинах МТ4. Каботегравір продемонстрував противірусну активність у клітинній культурі в панелі з 24 клінічних ізолятів ВІЛ-1 (по три в кожній групі M клад A, B, C, D, E, F і G та 3 у групі O), при цьому значення EC50 щодо ВІЛ-1 коливалися від 0,02 нМ до 1,06 нМ. Значення EC50 каботегравіру щодо трьох клінічних ізолятів ВІЛ-2 коливалися від 0,10 нМ до 0,14 нМ. Клінічних даних стосовно пацієнтів із ВІЛ-2 немає.
Противірусна активність у поєднанні з іншими противірусними засобами
У дослідженнях комбінацій in vitro каботегравір чинив слабку синергічну противірусну дію в поєднанні з нуклеозидними інгібіторами зворотної транскриптази (ламівудин, тенофовіру дизопроксилу фумарат, емтрицитабін) та адитивну дію в поєднанні з ненуклеозидним інгібітором зворотної транскриптази рилпівірином.
Вплив людської сироватки та сироваткових білків
Дослідження in vitro показали 408-кратний зсув IC50 каботегравіру в присутності 100 % людської сироватки (методом екстраполяції), а значення IC50, скориговане з урахуванням зв’язування з білками (PA-IC50), у клітинах MT4 становило 102 нМ.
Резистентність in vitro
Виділення з ВІЛ-1 дикого типу та активність проти резистентних штамів: протягом 112-денного перегляду штаму IIIB не спостерігалися віруси з більше ніж 10-кратним підвищенням
EC50 каботегравіру. Подальші мутації інтегрази виникли після пасажу ВІЛ-1 дикого типу (з поліморфізмом T124A) у присутності каботегравіру: Q146L (діапазон зміни кратності 1,3–4,6), S153Y (діапазон зміни кратності 2,8–8,4) та I162M (зміна кратності = 2,8). Як зазначалося вище, виявлення T124A є відбором передіснуючого міноритарного варіанту, який не має диференціальної чутливості до каботегравіру. До 56-го дня не було виділено амінокислотних замін у ділянці інтегрази під час перегляду ВІЛ-1 дикого типу NL-432 у присутності каботегравіру в концентрації 6,4 нМ.
Серед відомих протестованих мутантних варіантів, резистентних до інтегрази, помірна резистентність (≥5 -кратна, але менше ніж 10-кратна) спостерігалася з боку штамів E92Q/N155H, G118R, G140S/Q148H, Y143H/N155H, Q148K, Q148R, T66K/L74M і G140S/Q148K. Висока резистентність (≥ 10-кратна) спостерігалася з боку штамів E138K/Q148K, V72I/E138K/Q148K, E138K/Q148R, E138K/G140S/Q148R, L74M/V75A/G140S/Q148H, G140C/Q148R, Q148R/N155H/ та G140 Q148R.
Резистентність in vivo
HPTN 083
При первинному аналізі в дослідженні HPTN 083 було зареєстровано 13 випадків інфікування в групі каботегравіру та 39 випадків у групі тенофовіру дизопроксилу фумарату (TDF)/емтрицитабіну (FTC). У групі каботегравіру протягом застосування ін’єкційного каботегравіру для ДКП спостерігалося 5 випадків інфікування, причому 4 учасники отримували ін’єкції вчасно, а 1 учасник отримав одну ін’єкцію поза графіком. П’ять випадків інфікування сталися через ≥ 6 місяців після останньої дози каботегравіру, застосованої для ДКП. Три випадки інфікування сталися в період пероральної підготовки.
Генотипування та фенотипування ВІЛ здійснювали під час першого візиту, коли вірусне навантаження ВІЛ становило > 500 копій/мл. З 13 випадків інфікування в групі каботегравіру 4 учасники мали мутації резистентності до інгібіторів переносу ланцюга інтегрази (ІПЛІ). У групі TDF/FTC серед 4 учасників із резистентністю до нуклеозидних інгібіторів зворотної транскриптази (НІЗТ) (в тому числі 3 із мультикласовою резистентністю) троє мали мутацію M184V/I, а один — K65R.
Жоден із 5 учасників, які були інфіковані після тривалої перерви в застосуванні каботегравіру, не мав мутацій резистентності до ІПЛІ. В одного з 5 учасників неможливо було здійснити ані генотипування, ані фенотипування, оскільки рівень РНК ВІЛ-1 становив лише 770 копій/мл. В одного з інших 4 учасників не вдалося визначити фенотип щодо інтегрази. У решти 3 учасників зберігалася сприйнятливість до всіх ІПЛІ.
Троє учасників інфікувалися в період пероральної підготовки перед ін’єкційним введенням каботегравіру. Один учасник із невизначеним рівнем каботегравіру в плазмі не мав мутацій резистентності до ІПЛІ та був сприйнятливим до всіх ІПЛІ. Два учасники з визначеними концентраціями каботегравіру в плазмі мали мутації резистентності до ІПЛІ. Перший учасник мав мутації резистентності до ІПЛІ E138E/K, G140G/S, Q148R та E157Q. Фенотип щодо інтегрази встановити не вдалося. Другий учасник мав мутації резистентності до ІПЛІ E138A та Q148R. Цей вірус був резистентним до каботегравіру (кратність зміни 5,92), але чутливим до долутегравіру (кратність зміни 1,69).
П’ятеро учасників заразилися ВІЛ-1, незважаючи на ін’єкції каботегравіру, які 4 учасникам було введено вчасно, а одному — поза графіком. У двох учасників вірусне навантаження було занадто низьким для аналізу. Третій учасник не мав мутацій резистентності до ІПЛІ під час першого візиту з вірусемією (17-й тиждень), але мав мутацію R263K пізніше (станом на 112-й та 117-й дні). Хоча фенотип на 112-й день визначити не вдалося, фенотипування на 117-й день показало, що цей вірус є чутливим як до каботегравіру (кратність зміни 2,32), так і до долутегравіру (кратність зміни 2,29). Четвертий учасник мав мутації резистентності до ІПЛІ G140A та Q148R. Фенотипування показало резистентність до каботегравіру (кратність зміни 13), але чутливість до долутегравіру (кратність зміни 2,09). П’ятий учасник не мав мутацій резистентності до ІПЛІ.
Окрім 13 нових випадків інфікування, ще один учасник уже був інфікований ВІЛ-1 на момент включення в дослідження та не мав мутацій резистентності до ІПЛІ, однак через 60 днів були виявлені мутації резистентності до ІПЛІ E138K та Q148K. Встановити фенотип не вдалося.
Після первинного аналізу було проведено розширене ретроспективне вірусологічне тестування, щоб краще охарактеризувати часові межі інфікування ВІЛ. У результаті було встановлено, що з 13 інфікованих учасників, які вчасно отримували ін’єкції каботегравіру, один уже був інфікований раніше.
HPTN 084
При первинному аналізі в дослідженні HPTN 084 було зареєстровано 4 випадки інфікування в групі каботегравіру та 36 випадків інфікування в групі тенофовіру дизопроксилу фумарату (TDF)/емтрицитабіну (FTC).
У групі каботегравіру 2 випадки інфікування відбулися в період отримання ін’єкцій; одному учаснику 3 ін’єкції каботегравіру було зроблено із затримкою, причому обидва учасники не дотримувалися схеми перорального прийому каботегравіру.
Два випадки інфікування сталися після прийому останньої дози перорального каботегравіру; обидва учасники не дотримувалися схеми перорального прийому препарату. Перший візит із ВІЛ-позитивним результатом аналізу в одного учасника відбувся приблизно через 11 тижнів після включення в дослідження, в іншого — через 57 тижнів після включення в дослідження.
Генотипування ВІЛ здійснювали під час першого візиту, коли вірусне навантаження ВІЛ становило > 500 копій/мл (перший візит із вірусемією). Результати генотипування ВІЛ були наявні у 3 із 4 учасників групи каботегравіру. Значущих мутацій резистентності до ІПЛІ виявлено не було.
Результати генотипування ВІЛ були наявні у 33 із 36 учасників групи TDF/FTC, у яких сталося інфікування. Один учасник мав суттєву мутацію резистентності до НІЗТ (M184V); цей учасник також був резистентним до ННІЗТ, оскільки мав мутацію K103N. Дев’ять інших учасників були резистентними до ННІЗТ (у 7 була мутація K103N, окремо або в поєднанні з E138A чи P225H; у 1 була лише мутація K101E; 1 мав лише мутацію E138K).
Після первинного аналізу було проведено розширене ретроспективне вірусологічне тестування, щоб краще охарактеризувати часові межі інфікування ВІЛ. У результаті було встановлено, що з 4 інфікованих учасників, які отримували каботегравір, один уже був інфікований раніше.
Вплив на електрокардіограму
У рандомізованому плацебо-контрольованому перехресному дослідженні з трьома періодами 42 здорових добровольці були рандомізовані на 6 випадкових послідовностей та отримували три пероральні дози плацебо або каботегравіру по 150 мг кожні 12 годин (середнє значення Cmax в рівноважному стані було приблизно 2,8-кратно та 5,6-кратно вищим, ніж при пероральному прийомі каботегравіру 30 мг один раз на добу та ін’єкції каботегравіру 600 мг кожні 2 місяці відповідно) або одноразову дозу моксифлоксацину 400 мг (активний контроль). Після вихідного рівня та з поправкою на плацебо максимальна зміна середнього інтервалу QTc, узгоджена за часом, за методом корекції Фрідерічі (QTcF) при застосуванні каботегравіру становила 2,62 мсек (верхня межа 1-стороннього 90 % довірчого інтервалу [ДІ]: 5,26 мсек). Каботегравір не подовжував інтервал QTc протягом 24 годин після введення дози.
Клінічні дослідження
Клінічна ефективність
Ефективність АПРЕТЮДУ для зниження ризику інфікування ВІЛ-1 оцінювали у двох рандомізованих (1:1) подвійно сліпих багатоцентрових двогрупових контрольованих дослідженнях: HPTN 083 за участю чоловіків, не інфікованих ВІЛ-1, і трансгендерних жінок, які мають статеві контакти з чоловіками та у яких наявні ознаки високого ризику інфікування ВІЛ-1, а також HPTN 084 за участю цисгендерних жінок, які не інфіковані ВІЛ-1, але мають ризик інфікування. Ефективність АПРЕТЮДУ оцінювали в порівнянні з ефективністю щоденно перорально застосовуваного тенофовіру дизопроксилу фумарату (TDF)/емтрицитабіну (FTC).
Учасники, рандомізовані до групи АПРЕТЮДУ, спочатку приймали перорально одну таблетку АПРЕТЮДУ 30 мг 1 раз на добу та препарат плацебо протягом приблизно 5 тижнів, а надалі отримували внутрішньом’язові ін’єкції АПРЕТЮДУ (одноразова ін’єкція 600 мг [3 мл] через 1 місяць, 2 місяці та кожні 2 місяці в подальшому), а також щоденно приймали таблетку плацебо. Учасники, рандомізовані до групи TDF/FTC, спочатку приймали перорально TDF 300 мг/FTC 200 мг і препарат плацебо протягом приблизно 5 тижнів, а надалі
щодня приймали перорально TDF 300 мг/FTC 200 мг, а також отримували внутрішньом’язові ін’єкції плацебо (3 мл 20 % ліпідної емульсії для ін’єкцій) через 1 місяць, 2 місяці та кожні 2 місяці в подальшому).
HPTN 083
У дослідженні HPTN 083 з оцінки не меншої ефективності 4566 цисгендерних чоловіків і трансгендерних жінок, які мають статеві контакти з чоловіками, були рандомізовані у співвідношенні 1:1 та отримували досліджуваний препарат каботегравір (n = 2281) або TDF/FTC (n = 2285) у сліпому режимі до тижня 153.
На вихідному рівні середній вік учасників становив 26 років, 12 % були трансгендерними жінками, 72 % не були представниками європеоїдної раси, 67 % мали вік < 30 років.
Первинною кінцевою точкою була частота випадків інфікування ВІЛ серед учасників, рандомізованих до перорального та ін’єкційного введення АПРЕТЮДУ, порівняно з такою в групі перорального прийому TDF/FTC (з поправкою на раннє припинення). Первинний аналіз продемонстрував вищу ефективність АПРЕТЮДУ порівняно з TDF/FTC: ризик інфікування ВІЛ знизився на 66 %; коефіцієнт ризику (95 % ДІ) становив 0,34 (0,18–0,62). Подальший аналіз показав, що в групі каботегравіру один з учасників уже був інфікований раніше, тобто зниження ризику інфікування становило 69 % порівняно з таким в групі TDF/FTC (див. таблицю 1).
Таблиця 1. Первинна кінцева точка ефективності: порівняння частоти інфікування ВІЛ на рандомізованому етапі дослідження HPTN 083 (модифікована популяція всіх рандомізованих учасників (mITT), розширене ретроспективне вірусологічне тестування)
|
Показник |
Каботегравір (N = 2 278) |
TDF/FDC (N = 2 281) |
P-значення щодо вищої ефективності |
|
Людино-роки |
3211 |
3193 |
|
|
Інфекції ВІЛ-1 (частота інфікування на 100 людино-років) |
12a (0,37) |
39 (1,22) |
|
|
Відношення ризиків (95% ДІ) |
0,31 (0,16; 0,58) |
P = 0,0003 |
|
a Після первинного аналізу було проведено розширене ретроспективне вірусологічне тестування, щоб краще охарактеризувати часові межі інфікування ВІЛ. У результаті було встановлено, що з 13 інфікованих учасників, які отримували АПРЕТЮД, один учасник уже був інфікований раніше. Початковий коефіцієнт ризику (95 % ДІ) в первинному аналізі становить 0,34 (0,18–0,62).
|
|
Каботегравір TDF/FTC |
|
Каботегравір TDF/FTC |
|
Тенофовіру дизопроксилу фумарат (TDF)/емтрицитабін (FTC) |
|
Каботегравір (CAB) |
|
Кількість учасників у групі ризику |
|
К-сть тижнів від рандомізації |
|
Сукупна кількість явищ |
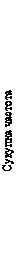
Рис. 1. Сукупна частота випадків інфікування ВІЛ у дослідженні HPTN 083
Результати аналізів у всіх підгрупах узгоджувалися із показником загального захисного ефекту. При цьому в учасників, рандомізованих до групи АПРЕТЮДУ, частота інфікування ВІЛ-1 була нижчою порівняно з такою в учасників, рандомізованих до групи TDF/FTC (див. таблицю 2).
Таблиця 2. Частота випадків інфікування ВІЛ у дослідженні HPTN 083 з розподілом за підгрупами (mITT, розширене ретроспективне вірусологічне тестування)
|
Підгрупа |
Частота інфікування у групі застосування каботегравіру на 100 людино-років |
Людино-років застосування препарату в групі каботегравіру |
Частота інфікування у групі застосування TDF/FTC на 100 людино-років |
Людино-років споживання препарату в групі TDF/FTC |
ВР (95 % ДІ) |
|
Вік |
|||||
|
< 30 років |
0,47 |
2110 |
1,66 |
1987 |
0,29 (0,15; 0,59) |
|
≥ 30 років |
0,18 |
1101 |
0,50 |
1206 |
0,39 (0,08; 1,84) |
|
Стать |
|||||
|
ЦГЧ |
0,35 |
2836 |
1,14 |
2803 |
0,32 (0,16; 0,64) |
|
ТГЖ |
0,54 |
371 |
1,80 |
389 |
0,34 (0,08; 1,56) |
|
Расова приналежність (США) |
|||||
|
Негроїдна раса |
0,58 |
691 |
2,28 |
703 |
0,26 (0,09; 0,76) |
|
Не негроїдна раса |
0,00 |
836 |
0,50 |
801 |
0,11 (0,00; 2,80) |
|
Регіон |
|||||
|
США |
0,26 |
1528 |
1,33 |
1504 |
0,21 (0,07; 0,60) |
|
Латинська Америка |
0,49 |
1020 |
1,09 |
1011 |
0,47 (0,17; 1,35) |
|
Азія |
0,35 |
570 |
1,03 |
581 |
0,39 (0,08; 1,82) |
|
Африка |
1,08 |
93 |
2,07 |
97 |
0,63 (0,06; 6,50) |
ЦГЧ — цисгендерні чоловіки, які мають статеві контакти з чоловіками.
ТГЖ — трансгендерні жінки, які мають статеві контакти з чоловіками.
HPTN 084
У дослідженні HPTN 084 з оцінки вищої ефективності 3224 цисгендерні жінки були рандомізовані у співвідношенні 1:1 та отримували досліджуваний препарат каботегравір (n = 1613) або TDF/FTC (n = 1610) у сліпому режимі до тижня 153.
На вихідному рівні середній вік учасників становив 25 років, > 99 % не були представниками європеоїдної раси, > 99 % складали цисгендерні жінки, 49 % мали вік < 25 років.
Первинною кінцевою точкою була частота інфікування ВІЛ серед учасників, рандомізованих для перорального та ін’єкційного введення АПРЕТЮДУ, порівняно з такою при пероральному прийомі TDF/FTC (з поправкою на раннє припинення). Первинний аналіз продемонстрував вищу ефективність АПРЕТЮДУ порівняно з TDF/FTC: ризик інфікування ВІЛ-1 знизився на 88 %; коефіцієнт ризику (95 % ДІ) становив 0,12 (0,05–0,31). Подальший аналіз показав, що в групі АПРЕТЮДУ один учасник уже був інфікований раніше, тобто зниження ризику інфікування ВІЛ-1 становило 90 % порівняно з таким в групі TDF/FTC (див. таблицю 3).
Таблиця 3. Первинна кінцева точка ефективності в дослідженні HPTN 084: порівняння частоти інфікування ВІЛ на рандомізованому етапі дослідження (mITT, розширене ретроспективне вірусологічне тестування)
|
Показник |
Каботегравір (N = 1613) |
TDF/FDC (N = 1610) |
P-значення щодо вищої ефективності |
|
Людино-роки |
1960 |
1946 |
|
|
Випадки інфікування ВІЛ-1 (частота інфікування на 100 людино-років) |
3a (0,15) |
36 (1,85) |
|
|
Відношення ризиків (95 % ДІ) |
0,10 (0,04; 0,27) |
P < 0,0001 |
|
a Після первинного аналізу було проведено розширене ретроспективне вірусологічне тестування, щоб краще охарактеризувати часові межі інфікування ВІЛ. У результаті було встановлено, що з 4 інфікованих учасників, які отримували АПРЕТЮД, один учасник уже був інфікований раніше. Початковий коефіцієнт ризику з поправкою на раннє припинення (95 % ДІ) у первинному аналізі становить 0,12 (0,05–0,31).
Рисунок 2. Сукупна частота випадків інфікування ВІЛ у дослідженні HPTN 084
|
|
Каботегравір TDF/FTC |
|
Каботегравір TDF/FTC |
|
Тенофовіру дизопроксилу фумарат (TDF)/емтрицитабін (FTC) |
|
Каботегравір (CAB) |
|
Кількість учасників у групі ризику |
|
К-сть тижнів від рандомізації |
|
Сукупна кількість явищ |
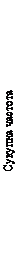
Рис. 2. Сукупна частота випадків інфікування ВІЛ у дослідженні HPTN 084
Результати заздалегідь запланованих аналізів у підгрупах узгоджувалися із показником загального захисного ефекту. При цьому в учасників, рандомізованих до групи АПРЕТЮДУ, частота інфікування ВІЛ-1 була нижчою порівняно з такою в учасників, рандомізованих до групи TDF/FTC (див. таблицю 4).
Таблиця 4. Частота інфікування ВІЛ-1 у дослідженні HPTN 084 з розподілом за підгрупами (mITT, розширене ретроспективне вірусологічне тестування)
|
Підгрупа |
Частота інфікування у групі каботегравіру на 100 людино-років |
Людино-років застосування препарату в групі каботегравіру |
Частота інфікування у групі TDF/FTC на 100 людино-років |
Людино-років застосування препарату в групі TDF/FTC |
Відношення ризиків (95 % ДІ) |
|
Вік |
|||||
|
< 25 років |
0,23 |
868 |
2,34 |
853 |
0,12 (0,03; 0,46) |
|
≥ 25 років |
0,09 |
1093 |
1,46 |
1093 |
0,09 (0,02; 0,49) |
|
Індекс маси тіла |
|||||
|
< 30 |
0,22 |
1385 |
1,88 |
1435 |
0,12 (0,04; 0,38) |
|
≥ 30 |
0,00 |
575 |
1,76 |
511 |
0,04 (0,00; 0,93) |
Діти віком від 12 років і з масою тіла не менше 35 кг
Безпека та ефективність застосування АПРЕТЮДУ для доконтактної профілактики ВІЛ-1 у дітей віком від 12 років з масою тіла не менше 35 кг, які належать до групи ризику, підтверджуються даними 2 відповідних і добре контрольованих досліджень застосування АПРЕТЮДУ для ДКП ВІЛ-1 у дорослих. Ці дані доповнюються даними щодо безпеки та фармакокінетики з досліджень за участю ВІЛ-1-інфікованих дорослих, які отримували препарат КАБЕНУВА (CABENUVA) (каботегравір і рилпівірин, суспензія для ін’єкцій пролонгованого вивільнення), а також за участю ВІЛ-1-інфікованих дітей, які отримували окремі компоненти препарату КАБЕНУВА додатково до поточної антиретровірусної терапії (див. розділи «Спосіб застосування та дози», «Побічні реакції», «Фармакокінетика»).
Фармакокінетика.
Фармакокінетика каботегравіру подібна у здорових і ВІЛ-інфікованих людей. Варіабельність фармакокінетики каботегравіру коливається від помірної до високої. У дослідженнях фази I за участю здорових добровольців коефіцієнт варіабельності (CVb%) значень AUC, Cmax і Ctau коливався від 34 % до 91 % в різних дослідженнях. Внутрішньосуб’єктна варіабельність (CVw%) є нижчою за міжсуб’єктну варіабельність.
Таблиця 5. Фармакокінетичні параметри каботегравіру після його перорального прийому 1 раз на добу та після початкових і подальших (кожні 2 місяці) внутрішньом’язових ін’єкцій
|
Етап введення |
Схема введення |
Середнє геометричне (5-й, 95-й процентиль)a |
||
|
AUC(0-tau)b (мк•год/мл) |
Cmax (мк/мл) |
Ctau (мк/мл) |
||
|
Пероральна підготовкаc |
30 мг 1 раз на добу |
145 (93,5; 224) |
8,0 (5,3; 11,9) |
4,6 (2,8; 7,5) |
|
Початкові ін’єкціїd |
600 мг в/м початкова доза |
1591 (714; 3245) |
8,0 (5,3; 11,9) |
1,5 (0,65; 2,9) |
|
Ін’єкції кожні 2 місяціe |
600 мг в/м 1 раз на 2 місяці |
3764 (2431; 5857) |
4,0 (2,3; 6,8) |
1,6 (0,8; 3,0) |
a Значення фармакокінетичних (ФК) параметрів ґрунтувалися на окремих ретроспективних оцінках популяційних ФК моделей для учасників досліджень фази III з оцінки лікування ВІЛ.
b tau – інтервал дозування: 24 години для перорального прийому; 1 місяць для початкових ін’єкцій та 2 місяці для подальших внутрішньом’язових ін’єкцій суспензії для ін’єкцій пролонгованої дії.
c Значення фармакокінетичних параметрів на етапі пероральної підготовки відповідають рівноважному стану.
d Значення Cmax на етапі початкових ін’єкцій переважно стосуються перорального прийому, оскільки початкова ін’єкція вводилася в той самий день, що й остання пероральна доза; однак значення AUC(0-tau) і Ctau відповідають початковій ін’єкції. У разі введення ВІЛ-інфікованим реципієнтам без пероральної підготовки (n = 110) середнє геометричне (5-й, 95-й процентиль) Cmax каботегравіру (через 1 тиждень після початкової ін’єкції) становило 1,89 мкг/мл (0,438–5,69), а Ctau — 1,43 мкг/мл (0,403–3,90).
e Значення фармакокінетичних параметрів відповідають рівноважному стану.
Абсорбція
Фармакокінетика ін’єкційного каботегравіру обмежена абсорбцією, оскільки каботегравір повільно всмоктується в системний кровотік із сідничного м’яза, що призводить до стійких концентрацій у плазмі крові. Після одноразової внутрішньом’язової ін’єкції 600 мг концентрація каботегравіру в плазмі піддається виявленню вже в перший день, при цьому концентрація каботегравіру через 4 години після введення становить у середньому 0,290 мг/мл (що перевищує значення PA-IC90 in vitro, яке дорівнює 0,166 мг/мл) та досягає максимальної плазмової концентрації з медіаною Tmax 7 днів. Цільові концентрації досягаються після початкової в/м ін’єкції (див. таблицю 10). Каботегравір виявляється у плазмі крові протягом 52 тижнів або довше після одноразової ін’єкції.
Після одноразової та багаторазових в/м ін’єкцій каботегравіру у дозі від 100 до 800 мг його концентрація в плазмі збільшується пропорційно або трохи менше, ніж пропорційно, до дози.
Розподіл
За даними in vitro каботегравір значною мірою (приблизно > 99 %) зв’язується з білками плазми крові людини. Після перорального прийому таблеток середній уявний об’єм розподілу (Vz/F) у плазмі становив 12,3 л. У людей оціночне значення Vc/F каботегравіру в плазмі становило 5,27 л, а Vp/F — 2,43 л. Ці оцінки об’єму разом із припущенням про високий рівень F дають підстави припустити, що каботегравір деякою мірою розподіляється в позаклітинному просторі.
Каботегравір присутній у жіночих і чоловічих статевих шляхах після одноразової внутрішньом’язової ін’єкції 3 мл (600 мг), що спостерігалося в дослідженні за участю здорових добровольців (n = 15). Середня концентрація каботегравіру на 3-й день (найбільш ранній зразок тканини для вивчення ФК) становила 0,49 мг/мл у тканині шийки матки, 0,29 мг/мл у цервіко-вагінальних виділеннях, 0,37 мг/мл у вагінальній тканині, 0,32 мг/мл у тканині прямої кишки та 0,69 мг/мл у ректальних виділеннях. Ці показники перевищують значення PA-IC90 in vitro.
Метаболізм
Каботегравір метаболізується переважно ферментом UGT1A1 з незначною участю UGT1A9. Каботегравір є основною циркулюючою сполукою в плазмі крові та складає > 90 % загальної дози міченого радіоактивним ізотопом препарату в плазмі. У людей після перорального прийому каботегравір виводиться переважно шляхом метаболізму; рівень виведення незміненого каботегравіру нирками дуже низький (< 1 % дози). Сорок сім відсотків загальної пероральної дози виводиться у вигляді незміненого каботегравіру з калом. Невідомо, повністю чи частково це пов’язано з неабсорбованим препаратом або екскрецією з жовчю кон’югату глюкуроніду, який може в подальшому розкладатися з утворенням вихідної сполуки в просвіті кишечнику. Каботегравір був присутній у зразках жовчі з дванадцятипалої кишки. Метаболіт у вигляді глюкуронової кислоти виявлявся в деяких, але не в усіх зразках жовчі з дванадцятипалої кишки. Двадцять сім відсотків загальної пероральної дози виводиться із сечею, переважно у вигляді глюкуроніду (75 % радіоактивного матеріалу в сечі, 20 % загальної дози).
Виведення
Середній кінцевий період напіввиведення каботегравіру становить 41 годину, а уявний кліренс (CL/F) — 0,21 л/годину, що спостерігалося після перорального прийому у здорових добровольців.
Особливі групи пацієнтів
Стать
Популяційні фармакокінетичні аналізи не виявили клінічно значущого впливу статі пацієнта на експозицію каботегравіру. Крім того, у дослідженні HPTN 083 не спостерігалося клінічно значущих відмінностей у плазмових концентраціях каботегравіру залежно від статі, у тому числі у цисгендерних чоловіків і трансгендерних жінок, які застосовували кросстатеву гормональну терапію, та у тих, хто її не застосовував. Тому корекція дози залежно від статі пацієнта не потрібна.
Расова приналежність
Популяційні фармакокінетичні аналізи не виявили клінічно значущого впливу расової приналежності пацієнта на експозицію каботегравіру, тому корекція дози залежно від расової приналежності не потрібна.
Індекс маси тіла
Популяційні фармакокінетичні аналізи не виявили клінічно значущого впливу ІМТ пацієнта на експозицію каботегравіру, тому корекція дози залежно від ІМТ не потрібна.
Діти (віком від >12 років до <18 років)
Популяційні фармакокінетичні аналізи не виявили клінічно значущих відмінностей в експозиції між ВІЛ-1-інфікованими дітьми та ВІЛ-1-інфікованими та не інфікованими дорослими учасниками досліджень каботегравіру, тому корекція дози для дітей з масою тіла ≥ 35 кг не потрібна.
Таблиця 6. Прогнозовані фармакокінетичні параметри після перорального прийому каботегравіру один раз на добу та після початкових внутрішньом’язових ін’єкцій і подальших ін’єкцій кожні 2 місяці у дітей віком від 12 до 18 років (≥ 35 кг)
|
Етап введення |
Схема введення |
Середнє геометричне (5-й, 95-й процентиль)a |
||
|
AUC(0-tau)b (мкг•год/мл) |
Cmax (мкг/мл) |
Ctau (мкг/мл) |
||
|
Пероральна підготовкаc |
30 мг один раз на добу |
193 (106; 346) |
14,4 (8,02; 25,5) |
5,79 (2,48; 12,6) |
|
Початкові ін’єкціїd |
600 мг в/м початкова доза |
2123 (881; 4938) |
11,2 (5,63; 21,5) |
1,84 (0,64; 4,52) |
|
Ін’єкції кожні 2 місяціe |
600 мг в/м раз на 2 місяці |
4871 (2827; 8232) |
7,23 (3,76; 14,1) |
2,01 (0,64; 4,73) |
a Значення фармакокінетичних (ФК) параметрів ґрунтувалися на популяційному моделюванні ФК у віртуальній популяції дітей, інфікованих ВІЛ-1, з масою тіла від 35 до 156 кг.
f tau – інтервал дозування: 24 години для перорального прийому; 1 місяць для початкових ін’єкцій та 2 місяці для подальших внутрішньом’язових ін’єкцій суспензії для ін’єкцій пролонгованої дії.
b Значення фармакокінетичних параметрів на етапі пероральної підготовки відповідають рівноважному стану.
c Значення Cmax на етапі початкових ін’єкцій переважно стосуються перорального прийому, оскільки початкова ін’єкція вводилася в той самий день, що й остання пероральна доза; однак значення AUC(0-tau) і Ctau відповідають початковій ін’єкції.
d Значення фармакокінетичних параметрів відповідають рівноважному стану.
Пацієнти літнього віку (> 65 років)
Популяційний фармакокінетичний аналіз не виявив клінічно значущого впливу віку пацієнта експозицію каботегравіру.
Даних щодо фармакокінетики каботегравіру в осіб віком понад 65 років недостатньо.
Порушення функції нирок
Не спостерігалося клінічно значущих фармакокінетичних відмінностей між пацієнтами з тяжкою нирковою недостатністю (які мають кліренс креатиніну < 30 мл/хв та не перебувають на діалізі) в порівнянні зі здоровими добровольцями. Корекція дози для осіб із порушенням функції нирок від легкого до тяжкого ступеня (які не перебувають на діалізі) не потрібна. Дослідження застосування каботегравіру пацієнтам, які перебувають на діалізі, не проводилися.
Порушення функції печінки
Не спостерігалося клінічно значущих фармакокінетичних відмінностей між пацієнтами з порушенням функції печінки середнього ступеня в порівнянні зі здоровими добровольцями. Пацієнтам із порушенням функції печінки легкого або середнього ступеня (ступінь А або В за класифікацією Чайлда – П’ю) корекція дози не потрібна. Вплив тяжкої печінкової недостатності (ступінь C за класифікацією Чайлда – П’ю) на фармакокінетику каботегравіру не досліджувався.
Особи, інфіковані вірусом гепатиту B та C
Немає даних щодо застосування каботегравіру особам, інфікованим ВГВ та ВГС, у дослідженнях доконтактної профілактики.
Поліморфізм ферментів, які метаболізують лікарські засоби
Метааналіз досліджень за участю здорових і ВІЛ-інфікованих осіб показав, що у ВІЛ-інфікованих пацієнтів із генотипами UGT1A1, що зумовлюють поганий метаболізм каботегравіру, спостерігалося в середньому 1,2-кратне підвищення показників AUC, Cmax і Ctau каботегравіру в рівноважному стані після ін’єкції порівняно з підвищенням в середньому в 1,38 раза після перорального прийому каботегравіру. Це було аналогічно 1,3–1,5-кратному середньому підвищенню значень AUC, Cmax і Ctau каботегравіру в рівноважному стані, яке спостерігалося після перорального прийому каботегравіру у здорових та ВІЛ-інфікованих осіб в сукупності. Ці відмінності не вважаються клінічно значущими. Поліморфізми ферменту UGT1A9 не були асоційовані з відмінностями у фармакокінетиці каботегравіру, тому корекція дози для людей із поліморфізмами ферментів UGT1A1 або UGT1A9 не потрібна.
Діти
Фармакокінетика та рекомендації щодо дозування каботегравіру у дітей віком до 12 років або з масою тіла ≤ 35 кг не встановлені.
Клінічні характеристики.
Показання.
АПРЕТЮД показаний дорослим і дітям (віком від 12 років) з масою тіла не менше 35 кг, які належать до групи ризику, для доконтактної профілактики (ДКП) з метою зниження ризику інфікування ВІЛ-1 статевим шляхом.
Перед початком застосування Апретюду для ДКП ВІЛ-1 необхідно мати задокументований негативний результат тесту на ВІЛ-1.
Протипоказання.
АПРЕТЮД протипоказаний:
• особам із невстановленим або позитивним статусом ВІЛ-1;
• особам із відомою гіперчутливістю до каботегравіру або до будь-якої з допоміжних речовин у таблетованій чи ін’єкційній лікарській формі препарату;
• особам, які приймають рифампіцин, рифапентин, фенітоїн, фенобарбітал, карбамазепін або окскарбазепін.
Взаємодія з іншими лікарськими засобами та інші види взаємодій.
Вплив інших лікарських засобів на фармакокінетику каботегравіру
Каботегравір метаболізується переважно уридиндифосфатглюкуронозилтрансферазою (UGT) 1A1 за деякою участю UGT1A9. Очікується, що лікарські засоби, які є сильними індукторами UGT1A1 або UGT1A9, знижуватимуть концентрацію каботегравіру в плазмі, що призведе до недостатньої ефективності (див. розділ «Протипоказання»).
Дослідження із застосуванням фізіологічних фармакокінетичних моделей (ФФКМ) свідчать, що при одночасному застосуванні каботегравіру з препаратами, які інгібують ферменти UGT, клінічно значущої взаємодії не очікується.
In vitro каботегравір не був субстратом поліпептидів, що транспортують органічні аніони (OATP) 1B1, OATP1B3, OATP2B1, або транспортера органічних катіонів (OCT1).
Каботегравір є субстратом P-глікопротеїну (P-gp) і білка резистентності раку молочної залози (BCRP), однак через його високу проникність не очікується змін у всмоктуванні при сумісному застосуванні з інгібіторами P-gp або BCRP.
Вплив каботегравіру на фармакокінетику інших лікарських засобів
In vivo каботегравір не впливав на мідазолам, який є зондом для (CYP) 3A4 цитохрому P450. Каботегравір не є клінічно значущим інгібітором таких ферментів і транспортерів: CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B4, UGT2B7, UGT2B15, UGT2B17, P-gp, BCRP, експортуюча помпа жовчних кислот (BSEP), OCT1, OCT2, OATP1B1, OATP1B3, транспортер
білків екструзії лікарських засобів і токсинів (MATE) 1, MATE 2-K, білок множинної лікарської резистентності (MRP) 2 та MRP4.
Каботегравір інгібував транспортери органічних аніонів (OAT) 1 (IC50 = 0,81 мкМ) та OAT3 (IC50 = 0,41 мкМ) in vitro, однак, враховуючи результати моделювання із застосуванням ФФКМ, не очікується взаємодії із субстратами OAT у клінічно значущих концентраціях.
In vitro каботегравір не індукував ферменти CYP1A2, CYP2B6 та CYP3A4.
З огляду на наведені дані і результати досліджень взаємодії між лікарськими засобами не очікується, що каботегравір впливатиме на фармакокінетику препаратів, які є субстратами перелічених ферментів і транспортерів.
Зважаючи на профіль міжлікарської взаємодії in vitro та в клінічних умовах, не очікується, що каботегравір змінюватиме концентрації інших антиретровірусних препаратів, зокрема інгібіторів протеази, нуклеозидних інгібіторів зворотної транскриптази, ненуклеозидних інгібіторів зворотної транскриптази, інгібіторів інтегрази, інгібіторів проникнення та ібалізумабу.
Дослідження взаємодії ін’єкційного каботегравіру з іншими лікарськими засобами не проводились. Дані про взаємодію між лікарськими засобами, наведені в таблиці 7, були отримані під час досліджень перорального каботегравіру.
Таблиця 7. Взаємодії між лікарськими засобами
|
Клас супутнього препарату: назва препарату |
Вплив на концентрацію каботегравіру або супутнього препарату |
Клінічний коментар |
|
Ненуклеозидний інгібітор зворотної транскриптази: Етравірин |
Каботегравір « AUC 1 % Cmax 4 % CТ « 0 % |
Етравірин суттєво не змінював концентрацію каботегравіру в плазмі крові. Корекція дози не потрібна. |
|
Ненуклеозидний інгібітор зворотної транскриптази: Рилпівірин |
Каботегравір « AUC 12 % Cmax 5 % CТ 14 % Рилпівірин « AUC ¯ 1 % Cmax ¯ 4 % CТ ¯ 8 % |
Рилпівірин суттєво не змінював мірою концентрацію каботегравіру в плазмі крові та навпаки. У разі сумісного застосування корекція дози АПРЕТЮДУ або рилпівірину не потрібна. |
|
Рифампіцин |
Каботегравір ¯ AUC ¯ 59 % Cmax ¯ 6 % |
Рифампіцин суттєво знижував концентрацію каботегравіру в плазмі, що, ймовірно, призведе до втрати терапевтичного ефекту. Одночасне застосування АПРЕТЮДУ з рифампіцином протипоказане. Рекомендації щодо дозування при одночасному застосуванні АПРЕТЮДУ (перорального та ін’єкційного) з рифампіцином не встановлені. |
|
Рифапентин |
Каботегравір ¯ |
Рифапентин може суттєво знизити концентрацію каботегравіру в плазмі, одночасне застосування протипоказане. |
|
Рифабутин |
Каботегравір ¯ AUC ¯ 21 % Cmax ¯ 17 % CТ ¯ 8 % |
Таблетки АПРЕТЮД: Рифабутин суттєво не змінював концентрацію каботегравіру в плазмі крові. Корекція дози не потрібна. Суспензія для ін’єкцій АПРЕТЮД: Якщо прийом рифабутину розпочато раніше або одночасно з першою початковою ін’єкцією АПРЕТЮДУ, рекомендується така схема введення АПРЕТЮДУ: одна ін’єкція 3 мл (600 мг), через 2 тижні після якої виконується друга початкова ін’єкція 3 мл (600 мг), надалі 1 раз на місяць протягом прийому рифабутину. Якщо прийом рифабутину розпочато під час другої початкової ін’єкції або пізніше, рекомендованою схемою є введення 3 мл (600 мг) 1 раз на місяць протягом прийому рифабутину. Після припинення прийому рифабутину рекомендованою схемою застосування АПРЕТЮДУ є введення 3 мл (600 мг) 1 раз на 2 місяці. |
|
Протисудомні препарати: Карбамазепін Окскарбазепін Фенітоїн Фенобарбітал |
Каботегравір ¯ |
Індуктори метаболізму можуть суттєво знижувати концентрацію каботегравіру в плазмі крові. Одночасне застосування протипоказане. |
|
Антациди (наприклад магній, кальцій або алюміній) |
Каботегравір ¯ |
Таблетки АПРЕТЮД: Одночасне застосування антацидних добавок потенційно може зменшити всмоктування каботегравіру при пероральному прийомі, але це питання не досліджувалося. Антацидні препарати, які містять полівалентні катіони, рекомендується застосовувати щонайменше за 2 години до або через 4 години після перорального прийому каботегравіру. Суспензія для ін’єкцій АПРЕТЮД: При парентеральному введенні взаємодія не є значущою. |
|
Пероральні контрацептиви (етинілестрадіол (EE) та левоноргестрел) |
EE « AUC 2 % Cmax ¯ 8 % CТ « 0 % LNG « |
Каботегравір не змінював концентрації етинілестрадіолу та левоноргестрелу в плазмі до клінічно значущого ступеня. У разі одночасного застосування з АПРЕТЮДОМ корекція дози пероральних контрацептивних засобів не потрібна. |
Особливості застосування.
Загальна стратегія запобігання інфікуванню ВІЛ-1
АПРЕТЮД не завжди є ефективним для профілактики зараження ВІЛ-1 (див. розділ «Фармакологічні властивості»). Час до настання захисного ефекту після початку застосування АПРЕТЮДУ не встановлений.
АПРЕТЮД використовують для доконтактної профілактики в рамках загальної стратегії запобігання інфікуванню ВІЛ-1, яка включає також застосування інших заходів профілактики ВІЛ-1 (наприклад, знання статусу ВІЛ-1, регулярне тестування на інші інфекції, що передаються статевим шляхом, користування презервативами).
АПРЕТЮД слід застосовувати для зниження ризику зараження ВІЛ-1 в осіб, у яких підтверджено негативний статус ВІЛ (див. розділ «Протипоказання»). Протягом періоду прийому АПРЕТЮДУ для доконтактної профілактики необхідно періодично підтверджувати в людини негативний статус ВІЛ (відповідно до місцевих рекомендацій, але не рідше ніж один раз на 3 місяці).
Для дітей можуть бути корисними більш часті візити та консультації, щоб забезпечити дотримання графіка введення препарату та тестування.
Якщо з’являються клінічні симптоми, характерні для гострої вірусної інфекції, та є підозра на нещодавнє (< 1 місяця) зараження ВІЛ-1, статус ВІЛ-1 слід підтвердити повторно.
Потенційний ризик резистентності
Існує потенційний ризик розвитку резистентності до АПРЕТЮДУ, якщо людина інфікується ВІЛ-1 до або під час застосування АПРЕТЮДУ чи після припинення прийому препарату (див. «Пролонгована дія ін’єкційного АПРЕТЮДУ»).
Щоб мінімізувати цей ризик, важливо періодично проводити клінічне обстеження щодо ризику зараження ВІЛ і часто робити аналізи для підтвердження ВІЛ-негативного статусу. Особам, у яких є підозрюване або підтверджене зараження ВІЛ-1, необхідно негайно розпочати антиретровірусну терапію (АРТ).
Для осіб, у яких ризик інфікування ВІЛ є постійним, після припинення застосування АПРЕТЮДУ слід розглянути альтернативні форми ДКП і розпочати їх протягом 2 місяців після останньої ін’єкції АПРЕТЮДУ.
Пролонгована дія ін’єкційного АПРЕТЮДУ
Залишкові концентрації ін’єкційного каботегравіру можуть зберігатися в системному кровообігу людини протягом тривалого періоду часу (до 12 місяців або довше), тому лікарі повинні брати до уваги пролонговану дію АПРЕТЮДУ після припинення його застосування (див. розділи «Взаємодія з іншими лікарськими засобами та інші види взаємодії», «Застосування у період вагітності або годування груддю», «Передозування»).
Важливість дотримання графіка
Необхідні періодичні консультації щодо важливості чіткого дотримання рекомендованого графіка введення АПРЕТЮДУ, щоб зменшити ризик інфікування ВІЛ-1 і потенційного розвитку резистентності.
Реакції гіперчутливості
Повідомлялося про реакції гіперчутливості при застосуванні інших інгібіторів інтегрази. Ці реакції характеризувалися висипом, конституційними змінами та іноді дисфункцією органів, в тому числі ураженням печінки. Пероральну підготовку за допомогою таблеток АПРЕТЮД використовували в клінічних дослідженнях, щоб ідентифікувати осіб, у яких може бути ризик виникнення реакцій гіперчутливості. Хоча на сьогоднішній день не спостерігалося жодних подібних реакцій при прийомі АПРЕТЮДУ, лікарям слід бути пильними та негайно припиняти застосування АПРЕТЮДУ та інших підозрюваних препаратів у разі появи ознак або симптомів гіперчутливості (таких як, зокрема, висип тяжкого ступеня або висип, що супроводжується лихоманкою, загальне нездужання, втома, біль у м’язах або суглобах, поява пухирів, ураження ротової порожнини, кон’юнктивіт, набряк обличчя, гепатит, еозинофілія або ангіоневротичний набряк). Необхідно контролювати клінічний стан, в тому числі рівень печінкових амінотрансфераз, і розпочати відповідну терапію (див. розділи «Спосіб застосування та дози», «Протипоказання», та підрозділ «Клінічні дослідження» розділу «Фармакологічні властивості»).
Гепатотоксичність
Повідомлялося про випадки гепатотоксичності при прийомі АПРЕТЮДУ в обмеженої кількості осіб із наявним захворюванням печінки або без такого (див. розділ «Побічні реакції»).
У разі підтвердження гепатотоксичності слід припинити застосування АПРЕТЮДУ, розглянути можливість клінічного та лабораторного моніторингу та надати лікування відповідно до клінічних показань (див. підрозділ «Пролонгована дія ін’єкційного АПРЕТЮДУ»).
Взаємодія з лікарськими засобами
Слід з обережністю призначати АПРЕТЮД одночасно з лікарськими засобами, які можуть знижувати його експозицію (див. розділ «Взаємодія з іншими лікарськими засобами та інші види взаємодій»).
Застосування в разі порушення функції печінки
Див. розділ «Спосіб застосування та дози» і підрозділи «Фармакокінетика» та «Особливі групи пацієнтів» розділу «Фармакологічні властивості».
Застосування в разі порушення функції нирок
Див. розділ «Спосіб застосування та дози» і підрозділи «Фармакокінетика» та «Особливі групи пацієнтів» розділу «Фармакологічні властивості».
Застосування особам літнього віку
Див. розділ «Спосіб застосування та дози» і підрозділи «Фармакокінетика» та «Особливі групи пацієнтів» розділу «Фармакологічні властивості».
Діти
Повідомлялося про суїцидальні думки та спроби суїциду при застосуванні каботегравіру, особливо у пацієнтів із наявними психічними захворюваннями (див. розділ «Побічні реакції»). Хоча клінічні дослідження не показали підвищеної частоти психічних захворювань у дітей порівняно з дорослими, враховуючи вразливість дитячої популяції, дітей слід консультувати перед призначенням і періодично під час прийому препарату АПРЕТЮД, а також лікувати відповідно до клінічних показань.
Допоміжні речовини
Особам із рідкісними спадковими захворюваннями непереносимості галактози, загальною недостатністю лактази або порушенням всмоктування глюкози-галактози не слід приймати АПРЕТЮД.
АПРЕТЮД містить менше 1 ммоль натрію (23 мг) на таблетку, тобто практично не містить натрію.
Застосування у період вагітності або годування груддю.
Фертильність. Немає даних про вплив каботегравіру на фертильність чоловіків або жінок. Дослідження на тваринах вказують на відсутність впливу каботегравіру на фертильність самців та самиць.
Каботегравір при пероральному введенні самцям і самицям щурів протягом 26 тижнів у дозі 1000 мг/кг/добу (що більше ніж у 30 разів перевищує експозицію у людини при максимальній рекомендованій дозі для людини [МРДЛ], яка становить 30 мг перорально або 400 мг внутрішньом’язово (в/м)) не чинив негативного впливу на репродуктивні органи самців і самиць, а також на сперматогенез. У щурів, які отримували каботегравір у дозах до 1000 мг/кг/добу, не спостерігалося функціонального впливу на спаровування або фертильність у самців і самиць.
Вагітність (категорія B1 згідно з класифікацією препаратів за ризиком для плода)
Даних щодо застосування каботегравіру вагітним жінкам недостатньо. Вплив АПРЕТЮДУ на вагітність у жінок не встановлений.
АПРЕТЮД під час вагітності можна застосовувати лише в тому випадку, якщо очікувана користь перевищує потенційний ризик для плода.
Каботегравір виявляється в системному кровообігу протягом 12 місяців після ін’єкції або навіть довше, тому слід брати до уваги можливість впливу на плід у період вагітності (див. розділ «Особливості застосування»).
У вагітних самиць щурів каботегравір проникав через плаценту і міг бути виявлений у тканинах плода. Каботегравір не чинив тератогенної дії у щурів при пероральних дозах до 1000 мг/кг/добу (що більше ніж у 30 разів перевищує експозицію у людей при МРДЛ, яка становить 30 мг перорально або 400 мг в/м), але спричиняв затримку пологів, що супроводжувалося зниженням виживаності та життєздатності потомства; у разі вилучення плода шляхом кесаревого розтину не відзначалося впливу на виживаність при народженні. Експозиції при застосуванні дози, яка не спричиняє видимих побічних ефектів (NOAEL), щонайменше в 11 разів перевищували експозицію у людей при введенні МРДЛ, яка становить 30 мг перорально або 400 мг в/м. Значущість цих даних для вагітності у жінок не встановлена.
Період годування груддю. На підставі результатів досліджень на тваринах очікується, що каботегравір буде виділятися в грудне молоко, хоча це не було підтверджено клінічними дослідженнями. Каботегравір може бути присутнім у грудному молоці жінки протягом 12 місяців або довше після останньої ін’єкції каботегравіру.
Грудне вигодовування може бути рекомендоване лише в тому випадку, якщо очікувана користь перевищує потенційний ризик для дитини.
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами.
Дослідження впливу каботегравіру на здатність керувати автотранспортом або працювати з іншими механізмами не проводилися. Оцінюючи здатність керувати автотранспортом або працювати з іншими механізмами, слід враховувати клінічний стан людини та профіль побічних ефектів АПРЕТЮДУ.
Спосіб застосування та дози.
Перш ніж розпочати застосування АПРЕТЮДУ, пацієнт повинен мати задокументований негативний результат тесту на ВІЛ-1 відповідно до чинних настанов.
Призначаючи АПРЕТЮД, медичні працівники повинні ретельно відбирати осіб, які погоджуються з графіком введення ін’єкцій, і консультувати їх щодо важливості дотримання запланованих візитів для введення препарату, щоб зменшити ризик зараження ВІЛ-1 (див. розділ «Особливості застосування»).
Дорослі та діти з масою тіла не менше 35 кг
Після бесіди з пацієнтом лікар може одразу зробити ін’єкцію АПРЕТЮДУ (див. рекомендації щодо введення препарату в таблиці 8).
В іншому варіанті можна застосувати таблетки АПРЕТЮД як підготовчий пероральний засіб для оцінки переносимості каботегравіру, перш ніж розпочати ін’єкційне введення АПРЕТЮДУ.
Початкові ін’єкції
Рекомендована початкова доза АПРЕТЮДУ — одноразова внутрішньом’язова ін’єкція 3 мл (600 мг). Якщо здійснювалась пероральна підготовка, першу ін’єкцію слід запланувати на останній день перорального прийому або протягом наступних 3 днів.
Через місяць слід виконати другу внутрішньом’язову ін’єкцію 3 мл (600 мг). Другу початкову ін’єкцію 3 мл (600 мг) можна робити за 7 днів до або через 7 днів після запланованої дати введення препарату.
Подальші ін’єкції
Після другої початкової ін’єкції рекомендована доза АПРЕТЮДУ становить 3 мл (600 мг) у вигляді одноразової внутрішньом’язової ін’єкції кожні 2 місяці. Ін’єкцію можна робити за 7 днів до або через 7 днів після запланованої дати введення препарату.
Таблиця 8. Рекомендована схема внутрішньом’язового введення
|
ПОЧАТКОВІ ІН’ЄКЦІЇ (з інтервалом один місяць) |
ПОДАЛЬШІ ІН’ЄКЦІЇ (з інтервалом два місяці) |
|
|
Препарат |
Безпосередньо ін’єкційне введення: місяці 1 і 2 або Після пероральної підготовки: місяці 2 і 3 |
Через два місяці після останньої початкової ін’єкції та надалі кожні 2 місяці |
|
Каботегравір |
3 мл (600 мг) |
3 мл (600 мг) |
Пропуск дози
Пропуск введення суспензії для ін’єкцій пролонгованої дії АПРЕТЮД
Наполегливо рекомендується чітко дотримуватись графіка виконання ін’єкцій.
Особи, які пропустили запланований візит для виконання ін’єкції, повинні повторно пройти клінічне обстеження та аналіз на ВІЛ, щоб переконатися, що відновлення ДКП залишається доцільним. Рекомендації щодо введення препарату після пропуску ін’єкції див. у таблиці 9.
У разі неможливості уникнути відхилення понад 7 днів від планового візиту для ін’єкції, можна приймати таблетки АПРЕТЮД (30 мг) один раз на добу замість одного запланованого візиту для ін’єкції.
Якщо пероральний прийом АПРЕТЮДУ для ДКП триває понад два місяці, рекомендується альтернативна схема.
Першу пероральну дозу для ДКП слід прийняти через два місяці (±7 днів) після останньої ін’єкції АПРЕТЮДУ. Відновлення ін’єкційного введення слід запланувати на останній день пероральної ДКП або протягом наступних 3 днів, як зазначено в таблиці 9.
Таблиця 9. Рекомендації щодо ін’єкційного введення після пропуску ін’єкцій або після прийому таблеток АПРЕТЮД замість ін’єкції
|
Пропущені дози |
|
|
Час від моменту останньої ін’єкції |
Рекомендація |
|
Якщо пропущено другу ін’єкцію, а після першої ін’єкції минуло: |
|
|
≤ 2 місяців |
Якнайшвидше зробіть одну ін’єкцію 3 мл (600 мг) і надалі здійснюйте введення препарату кожні 2 місяці. |
|
> 2 місяців |
Знову зробіть одну початкову ін’єкцію 3 мл (600 мг), а через місяць — другу початкову ін’єкцію 3 мл (600 мг). Далі дотримуйтесь графіка виконання ін’єкцій один раз на 2 місяці. |
|
Якщо пропущено третю або подальші ін’єкції, а після першої ін’єкції минуло: |
|
|
≤ 3 місяців |
Якнайшвидше зробіть одну ін’єкцію 3 мл (600 мг) і надалі здійснюйте введення препарату кожні 2 місяці. |
|
> 3 місяців |
Знову зробіть одну початкову ін’єкцію 3 мл (600 мг), а через місяць — другу початкову ін’єкцію 3 мл (600 мг). Далі дотримуйтесь графіка виконання ін’єкцій один раз на 2 місяці. |
Діти
Безпека та ефективність застосування АПРЕТЮДУ для дітей віком до 12 років із масою тіла менше 35 кг не встановлені.
Особливі групи пацієнтів
Пацієнти літнього віку
Пацієнтам літнього віку корекція дози не потрібна. Даних щодо застосування АПРЕТЮДУ особам віком ≥ 65 років недостатньо (див. підрозділи «Фармакокінетика» та «Особливі групи пацієнтів» розділу «Фармакологічні властивості»).
Пацієнти з порушенням функції нирок
Пацієнтам із порушенням функції нирок від легкого до тяжкого ступеня, які не перебувають на діалізі, корекція дози не потрібна (див. див. підрозділи «Фармакокінетика» та «Особливі групи пацієнтів» розділу «Фармакологічні властивості»).
Пацієнти з порушенням функції печінки
Пацієнтам із порушенням функції печінки легкого або середнього ступеня (ступінь А або В за класифікацією Чайлда – П’ю) корекція дози не потрібна. Застосування АПРЕТЮДУ пацієнтам із тяжкою печінковою недостатністю (ступінь C за класифікацією Чайлда – П’ю) не досліджувалось (див. підрозділи «Фармакокінетика» та «Особливі групи пацієнтів» розділу «Фармакологічні властивості»).
Спосіб застосування
Використовується лише для внутрішньом’язової (в/м) ін’єкції в сідничну ділянку. Не можна вводити внутрішньовенно.
Ін’єкцію препарату АПРЕТЮД має виконувати медичний працівник. Інструкції щодо використання препарату містяться в додатку «Інструкція із застосування».
У разі ін’єкційного введення АПРЕТЮДУ медпрацівники повинні брати до уваги індекс маси тіла (ІМТ) пацієнта, щоб бути впевненими, що голка має достатню довжину, щоб дістатися сідничного м’яза.
Діти
Безпека та ефективність застосування АПРЕТЮДУ для дітей віком до 12 років із масою тіла менше 35 кг не встановлені.
Передозування.
Симптоми та ознаки
Дотепер не зареєстровано випадків передозування препарату АПРЕТЮД.
Лікування
Спеціального лікування при передозуванні АПРЕТЮДУ не існує. У разі передозування слід призначити підтримувальну терапію з відповідним моніторингом, якщо це необхідно. Подальше лікування має надаватися відповідно до клінічних показань або до рекомендацій національного токсикологічного центру, якщо це можливо.
Відомо, що АПРЕТЮД значною мірою зв’язується з білками плазми; отже, діаліз навряд чи допоможе у виведенні препарату з організму. При лікуванні передозування ін’єкційного АПРЕТЮДУ слід брати до уваги тривалу дію препарату після ін’єкції (див. розділ «Особливості застосування»).
Побічні реакції.
Дані клінічних досліджень
Оцінка безпеки застосування АПРЕТЮДУ ґрунтується на двох клінічних дослідженнях фази III: HPTN 083 та HPTN 084. У дослідженні HPTN 083 середня тривалість застосування досліджуваного препарату в сліпому режимі становила 65 тижнів і 2 дні (від 1 дня до 156 тижнів і 1 дня), загальний показник застосування каботегравіру становив 3270 людино-років. У дослідженні HPTN 084 середня тривалість застосування досліджуваного препарату в сліпому режимі становила 64 тижні та 1 день (від 1 дня до 153 тижнів і 1 дня), загальний показник застосування каботегравіру становив 1920 людино-років.
Побічні реакції
Найчастіші побічні реакції, які реєструвалися більше ніж у 10 % учасників у кожній з експериментальних груп у дослідженнях HPTN 083 та HPTN 083, представлено в таблиці 10.
Таблиця 10. Найчастіші побічні реакції (зареєстровані у ≥ 10 % учасників у будь-якій експериментальній групі в дослідженнях HPTN 083 та HPTN 084)1 у термінах переважного використання
|
Побічна реакція |
HPTN 083 |
HPTN 084 |
||
|
Термін переважного використання |
Каботегравір (N = 2281) n (%) |
TDF/FTC (N = 2285) n (%) |
Каботегравір (N = 1613) n (%) |
TDF/FTC (N = 1610) n (%) |
|
Біль у місці ін’єкції |
1713 (75) |
688 (30) |
522 (32) |
147 (9) |
|
Знижений нирковий кліренс креатиніну |
1576 (69) |
1661 (73) |
1160 (72) |
1192 (74) |
|
Підвищений рівень креатинфосфокінази в крові |
506 (22) |
497 (22) |
237 (15) |
263 (16) |
|
Назофарингіт |
383 (17) |
379 (17) |
82 (5) |
96 (6) |
|
Підвищений рівень креатиніну в крові |
379 (17) |
426 (19) |
363 (22) |
347 (22) |
|
Головний біль |
377 (17) |
356 (16) |
377 (23) |
373 (23) |
|
Діарея |
328 (14) |
336 (15) |
101 (6) |
119 (7) |
|
Анальна хламідійна інфекція |
264 (12) |
297 (13) |
- |
- |
|
Інфекція верхніх дихальних шляхів |
264 (12) |
271 (12) |
268 (17) |
293 (18) |
|
Вузлик у місці ін’єкції |
263 (12) |
13 (< 1) |
80 (5) |
5 (< 1) |
|
Підвищений рівень ліпази |
255 (11) |
272 (12) |
198 (12) |
171 (11) |
|
Затвердіння в місці ін’єкції |
255 (11) |
8 (< 1) |
70 (4) |
4 (< 1) |
|
Підвищений рівень глюкози в крові |
247 (11) |
166 (7) |
584 (36) |
451 (28) |
|
Пірексія |
232 (10) |
112 (5) |
22 (1) |
21 (1) |
|
Гонококовий проктит |
220 (10) |
236 (10) |
- |
- |
|
Підвищений рівень аспартатамінотрансферази |
213 (9) |
220 (10) |
212 (13) |
181 (11) |
|
Підвищений рівень аланінамінотрансферази |
186 (8) |
220 (10) |
232 (14) |
228 (14) |
|
Підвищений рівень амілази |
158 (7) |
183 (8) |
558 (35) |
573 (36) |
|
Знижений рівень глюкози в крові |
109 (5) |
118 (5) |
425 (26) |
439 (27) |
|
Знижений рівень фосфору в крові |
107 (5) |
126 (6) |
278 (17) |
322 (20) |
|
Інфекції сечовивідних шляхів |
32 (1) |
23 (1) |
225 (14) |
210 (13) |
|
Дисфункціональні маткові кровотечі |
- |
- |
161 (10) |
161 (10) |
|
Вульвовагінальний кандидоз |
- |
- |
139 (9) |
162 (10) |
1 На етапі дослідження із застосуванням перорального та ін’єкційного препарату в сліпому режимі.
Побічні реакції наведено в порядку зменшення частоти їх виникнення згідно з даними, отриманими в групі каботегравіру в дослідженні HPTN 083.
Наведені побічні реакції включають ті, які можна віднести на рахунок застосування пероральних або ін’єкційних форм АПРЕТЮДУ. У разі відмінностей у частоті виникнення реакції між дослідженнями HPTN 083 та HPTN 084 зазначається найвища категорія частоти.
Найчастішими побічними реакціями в дослідженні HPTN 083 були реакції в місці ін’єкції (РМІ) (82 %), головний біль (17 %) і діарея (14 %).
Найчастішими побічними реакціями в дослідженні HPTN 084 були реакції в місці ін’єкції (38 %), головний біль (23 %) і підвищений рівень трансаміназ (19 %).
Побічні реакції, виявлені в цих дослідженнях, наведено нижче із зазначенням класу систем органів за Медичним словником для регуляторної діяльності (MedDRA) та частоти виникнення.
Частоту побічних реакцій визначено таким чином:
дуже часто (≥ 1/10);
часто (≥ 1/100, < 1/10);
нечасто (≥1/1000, < 1/100);
рідко (≥ 1/10000, < 1/1000);
дуже рідко (< 1/10000);
невідомо (неможливо оцінити за наявними даними).
Таблиця 11. Побічні реакції1
|
Клас систем органів (КСО) за MedDRA |
Категорія частоти |
Побічні реакції |
|
Психічні розлади |
Часто |
Аномальні сновидіння, безсоння, депресія |
|
Нечасто |
Спроба самогубства*, суїцидальні думки* (особливо в осіб з уже наявними психічними захворюваннями) |
|
|
Порушення з боку нервової системи |
Дуже часто |
Головний біль |
|
Часто |
Запаморочення |
|
|
Нечасто |
Вазовагальні реакції (у відповідь на ін’єкції) |
|
|
Порушення з боку шлунково-кишкового тракту |
Дуже часто |
Діарея |
|
Часто |
Нудота Біль у животі2 Метеоризм Блювання |
|
|
Порушення з боку гепатобіліарної системи |
Дуже часто |
Підвищений рівень трансаміназ |
|
Нечасто |
Гепатотоксичність |
|
|
Порушення з боку шкіри та підшкірної клітковини |
Часто |
Висип3 |
|
Нечасто |
Кропив’янка*, ангіоневротичний набряк* |
|
|
Порушення з боку скелетно-м’язової системи та сполучної тканини |
Часто |
Міалгія |
|
Загальні порушення та реакції в місці введення |
Дуже часто |
Пірексія4 Реакції в місці ін’єкції5 (біль і болісна чутливість, утворення вузликів, затвердіння) |
|
Часто |
Реакції в місці ін’єкції5 (набряк, синець, почервоніння, відчуття жару, свербіж, втрата чутливості) Втомлюваність Нездужання |
|
|
Нечасто |
Реакції в місці ін’єкції5 (гематома, знебарвлення, абсцес) |
|
|
Результати лабораторних та інструментальних досліджень |
Нечасто |
Збільшення маси тіла |
1 Частота виявлених побічних реакцій визначається на підставі всіх зареєстрованих явищ і не обмежується тими, які, на думку дослідника, можуть бути пов’язані із застосуванням досліджуваного препарату.
2 Біль у животі є груповим терміном переважного використання за MedDRA, який включає біль у верхній частині живота та біль у животі.
3 Висип є груповим терміном за MedDRA, який включає висип, еритематозний висип, макулярний висип, макулопапульозний висип, морбіліформний висип, папульозний висип, сверблячий висип.
4 Пірексія є груповим терміном за MedDRA, який включає лихоманку та відчуття жару. У більшості випадків пірексія спостерігалася протягом одного тижня після ін’єкції.
5 РМІ, наведені в таблиці, відзначалися у 2 або більше учасників.
* Цю побічну реакцію було виявлено в постмаркетингових звітах. Категорія частоти базується на побічних реакціях у осіб, які отримували каботегравір у рандомізованих клінічних дослідженнях.
Реакції у місці введення
У дослідженні HPTN 083 2 % учасників припинили застосування АПРЕТЮДУ через виникнення реакцій у місці ін’єкції (РМІ).
При проведенні 20286 ін’єкцій було зареєстровано 8900 РМІ.
Загалом 2117 учасників отримали принаймні одну ін’єкцію. Серед 1740 (82 %) учасників, у яких виникла хоча б одна РМІ, максимальна тяжкість РМІ була легкою (ступінь 1, 34 % учасників), помірною (ступінь 2, 46 % учасників) або важкою (ступінь 3, 3 % учасників). У жодного учасника не було РМІ 4-го ступеня. Середня тривалість усіх РМІ в сукупності становила 4 дні. Частка учасників, у яких спостерігалися РМІ під час кожного візиту, та тяжкість РМІ з часом зменшувалися.
У дослідженні HPTN 084 жоден з учасників не припинив застосування АПРЕТЮДУ через виникнення РМІ. При проведенні 13068 ін’єкцій було зареєстровано 1171 РМІ.
Загалом 1519 учасників отримали принаймні одну ін’єкцію. Серед 3578 (38 %) учасників, у яких виникла хоча б одна РМІ, максимальна тяжкість РМІ була легкою (ступінь 1, 25 % учасників), помірною (ступінь 2, 13 % учасників) або важкою (ступінь 3, < 1 % учасників). У жодного учасника не було РМІ 4-го ступеня. Середня тривалість усіх РМІ в сукупності становила 8 днів. Частка учасників, у яких спостерігалися РМІ під час кожного візиту, та тяжкість РМІ з часом зменшувалися.
Збільшення маси тіла
До 41-го тижня та до 97-го тижня дослідження HPTN 083 в учасників, які отримували АПРЕТЮД, маса тіла збільшилася в середньому на 1,2 кг (міжквартильний розмах [IQR] від –1,0 до 3,5; n = 1623) та на 2,1 кг (IQR від –0,9 до 5,9; n = 601) від вихідного рівня відповідно; учасники групи тенофовіру дизопроксилу фумарату (TDF)/емтрицитабіну (FTC) набрали в середньому 0,0 кг (IQR від –2,1 до 2,4; n = 1611) та 1,0 кг (IQR від –1,9 до 4,0; n = 598) від вихідного рівня відповідно.
До 41-го тижня та до 97-го тижня дослідження HPTN 084 в учасників, які отримували АПРЕТЮД, маса тіла збільшилася в середньому на 2,0 кг (IQR від 0,0 до 5,0; n = 1151) та на 4,0 кг (IQR від 0,0 до 8,0; n = 216) від вихідного рівня відповідно; учасники групи тенофовіру дизопроксилу фумарату (TDF)/емтрицитабіну (FTC) набрали в середньому 1,0 кг (IQR від –1,0 до 4,0; n = 1131) та 3,0 кг (IQR від –1,0 до 6,0; n = 218) від вихідного рівня відповідно.
Вплив на результати лабораторних аналізів
У дослідженнях HPTN 083 та HPTN 084 приблизно однакова кількість пацієнтів у групах застосування каботегравіру та тенофовіру дизопроксилу фумарату (TDF)/емтрицитабіну (FTC) мала підвищені рівні печінкових трансаміназ (АЛТ/АСТ), при цьому максимальне підвищення від вихідного рівня було переважно 1-го та 2-го ступеня. У дослідженні HPTN 083 кількість учасників з максимальним підвищенням показників АЛТ (3-го або 4-го ступеня) від вихідного рівня становила 40 (2 %) проти 44 (2 %) у групах каботегравіру та TDF/FTC, а підвищення рівня АСТ 3-го або 4-го ступеня відзначалося у 68 (3 %) та у 79 (3 %) учасників відповідно. У дослідженні HPTN 084 кількість учасників з максимальним підвищенням показників АЛТ (3-го або 4-го ступеня) від вихідного рівня становила 12 (< 1 %) проти 18 (1 %) у групах каботегравіру та TDF/FTC, а підвищення рівня АСТ 3-го або 4-го ступеня відзначалося у 15 (< 1 %) та у 14 (< 1 %) учасників відповідно.
У кількох учасників як у групі каботегравіру, так і в групі TDF/FTC спостерігалися побічні явища у вигляді підвищення рівнів АСТ або АЛТ, що призвело до припинення прийому досліджуваного препарату. У дослідженні HPTN 083 кількість учасників у групах каботегравіру та TDF/FTC, які припинили прийом препарату через підвищення рівня АЛТ, становила 29 (1 %) і 31 (1 %), а через підвищення рівня АСТ — 7 (< 1 %) і 8 (< 1 %) відповідно. У дослідженні HPTN 084 кількість учасників у групах каботегравіру та TDF/FTC, які припинили прийом лікарського засобу через підвищення рівня АЛТ, становила 12 (< 1 %) проти 15 (< 1 %). Випадків припинення прийому препарату через підвищення рівня АСТ не було.
Діти
У дітей, які отримували АПРЕТЮД для доконтактної профілактики ВІЛ-1, дані про безпеку були зіставні з даними про безпеку в дорослих, які отримували АПРЕТЮД для ДКП ВІЛ-1 (див. підрозділи «Фармакодинаміка» та «Клінічні дослідження» розділу «Фармакологічні властивості»).
На підставі даних двох відкритих багатоцентрових клінічних досліджень за участю 64 ВІЛ-неінфікованих дітей з групи ризику (віком до 18 років із масою тіла ≥ 35 кг на момент реєстрації), які отримували каботегравір, не було виявлено нових проблем щодо безпеки порівняно з безпекою, встановленою для дорослих, які отримували каботегравір для PrEP ВІЛ-1 у HPTN 083 та HPTN 084.
На підставі даних аналізу 16-го тижня дослідження MOCHA за участю 23 ВІЛ-інфікованих дітей (віком від 12 років з масою тіла ≥ 35 кг), які отримували базову комбіновану антиретровірусну терапію, не було виявлено жодних нових проблем щодо безпеки при додаванні перорального каботегравіру з подальшим ін’єкційним каботегравіром (n = 8) порівняно з профілем безпеки, встановленим для дорослих при застосуванні каботегравіру (див. підрозділ «Фармакодинаміка» розділу «Фармакологічні властивості»).
Звітування про підозрювані побічні реакції
Повідомлення про побічні реакції після реєстрації лікарського засобу має важливе значення. Це дає змогу проводити моніторинг співвідношення користь/ризик при застосуванні цього лікарського засобу. Медичним та фармацевтичним працівникам, а також пацієнтам або їхнім законним представникам слід повідомляти про усі випадки підозрюваних побічних реакцій та відсутності ефективності лікарського засобу через Автоматизовану інформаційну систему з фармаконагляду за посиланням: https://aisf.dec.gov.ua.
Термін придатності. 3 роки.
Умови зберігання. Зберігати при температурі не вище 30 °С. Не заморожувати. Зберігати у недоступному для дітей місці. Після набору в шприц препарат необхідно ввести протягом 2 годин.
Несумісність. Оскільки дослідження сумісності не проводились, каботегравір для ін’єкцій не можна змішувати з іншими лікарськими засобами.
Упаковка. Прозорий скляний флакон, закупорений бромбутиловою резиновою пробкою та алюмінієвим ковпачком зі знімною пластиковою кришечкою. По 3 мл у флаконі; по 1 або 25 флаконів в картонній коробці.
Категорія відпуску. За рецептом. Виробник. Глаксо Оперейшнс ЮК Лімітед, що веде діяльність як Глаксо Веллком Оперейшнс / Glaxo Operations UK Ltd (trading as Glaxo Wellcome Operations).Місцезнаходження виробника та його адреса місця провадження діяльності.
Хармір Роуд, Барнард Кастл, DL12 8DT, Велика Британія /
Harmire Road, Barnard Castle, DL12 8DT, United Kingdom
Заявник та/або представник заявника.
ТОВ «ГлаксоСмітКляйн Фармасьютікалс Україна».
Місцезнаходження заявника та/або представника заявника.
02152, м. Київ, проспект Павла Тичини, буд. 1-В, тел: (044) 585-51-85, факс: (044) 585-51-92.
Також про усі випадки підозрюваних побічних реакцій та відсутності ефективності лікарського засобу можна повідомляти ТОВ «ГлаксоСмітКляйн Фармасьютікалс Україна» за цілодобовим телефоном (044) 585-51-85 або на email [email protected].